|
Курсовая работа: Кинетические методы определения загрязнителей в различных природных средахКурсовая работа: Кинетические методы определения загрязнителей в различных природных средах«Кинетические методы определения загрязнителей в различных природных средах» Содержание Введение Глава 1.Сущность кинетических (ферментативных) методов анализа Глава 2. Некоторые основные сведения о ферментах 2. 1. Возможности ферментативных методов анализа 2. 2. Определение органических соединений 2. 3. Определение неорганических соединений Глава 3. Примеры использования кинетических методов в анализе объектов Глава 4. Оборудование для кинетических методов Литература ВВЕДЕНИЕ Для контроля примесей в объектах пищевой, микробиологической и фармацевтической промышленности, в мониторинге окружающей среды, для решения некоторых медицинских и биохимических задач в последние годы все шире применяют ферментативные методы анализа, основанные на использовании зависимости скорости катализируемой ферментом химической реакции от концентрации реагирующих веществ и фермента. Использование биологических катализаторов, отличающихся высокой активностью и избирательностью действия, позволяет значительно повысить чувствительность и селективность методов анализа. Эти качества в сочетании с простотой аппаратурного оформления и методики эксперимента, экспрессностью обосновывают широкое внедрение ферментативных методов в практику клинических, агрохимических, заводских лабораторий, научно-исследовательских институтов, природоохранных служб. Ферментативными методами можно определять сами ферменты, их субстраты (то есть соединения, превращение которых катализируют ферменты), а также соединения, воздействующие на каталитическую активность ферментов, - их эффекторы, активаторы или ингибиторы (то есть вещества, которые либо повышают, либо понижают активность фермента). ГЛАВА 1. СУЩНОСТЬ КИНЕТИЧЕСКИХ (ФЕРМЕНТАТИВНЫХ) МЕТОДОВ АНАЛИЗА Кинетические методы основаны на использовании зависимости скорости химической реакции от концентрации реагирующих веществ, а в случае каталитических реакций и от концентрации катализатора:
где v — скорость; K — константа скорости каталитической реакции; сА, сВ и скат концентрации реагирующих веществ и катализатора; m, n и р — показатели степени при концентрациях реагентов и катализатора (обычно р = 1). Аналитическим сигналом в кинетических методах является скорость процесса или пропорциональная ей величина. Реакцию, скорость которой измеряется, называют индикаторной, а вещество, по изменению концентрации которого судят о скорости процесса — индикаторным веществом. Индикаторные реакции могут быть основаны на катализе окислительно-восстановительных реакций, реакций замещения в координационной сфере ионов металлов, реакций гидролиза и декарбоксилирования органических соединений. Наиболее чувствительны и сравнительно просто выполнимы окислительно-восстановительные каталитические реакции. Они чаще всего используются в анализе неорганических веществ. Кроме каталитических реакций в кинетических методах используют и некаталитические реакции окисления-восстановления, разложения, осаждения. К индикаторной реакции предъявляют ряд требований: · концентрация определяемого компонента за время наблюдения практически не должна меняться. Катализатор в ходе реакции не расходуется. Если же определяемым является одно из реагирующих веществ (некаталитический вариант метода), то с достаточной точностью его можно определять в тот начальный период, когда его концентрация изменяется не более чем на 5%; · необходимо наличие быстрого, простого и доступного метода наблюдения за скоростью индикаторной реакции, т. е. за изменением концентрации индикаторного вещества во времени. · скорость индикаторной реакции должна находиться в определенных пределах. Оптимальное время наблюдения за скоростью индикаторной реакции 5–15 мин. Однако с развитием методов изучения быстрых процессов все чаще используют реакции, протекающие с большой скоростью. Существуют два варианта кинетических методов: В каталитическом варианте кинетического метода (каталитическом методе, каталиметрии) определяемый компонент или связанные с ним соединения являются катализатором индикаторной реакции. В некаталитическом варианте кинетического метода определяемым компонентом является одно из реагирующих веществ в некаталитической или каталитической индикаторной реакции. Каталитические методы отличаются от других химических методов анализа высокой чувствительностью, а некаталитический вариант кинетических методов – высокой селективностью. Возможны различные способы определения неизвестной концентрации вещества по данным кинетических измерений. Если индикаторным веществом является продукт реакции, и его текущую концентрацию обозначить через х, то скорость реакции можно выразить как
На начальной стадии реакции концентрации определяемого вещества В и реагента А могут практически не изменяться за время наблюдения за скоростью процесса. Тогда, проинтегрировав уравнение (2), получим
т. е. наблюдается линейная зависимость между концентрацией индикаторного вещества и временем. Кинетический метод, основанный на использовании этого уравнения, называют дифференциальным. Если концентрация хотя бы одного из реагирующих веществ за время наблюдения за скоростью реакции заметно меняется (более чем на 10%), то между концентрацией индикаторного вещества и временем существует более сложная (например, логарифмическая, обратная и т. д.) зависимость. Такой кинетический метод называют интегральным. В интегральном варианте часто применяют построение зависимостей концентрации индикаторного вещества от времени в полулогарифмических, обратных или других координатах, т. к. для расчета неизвестной концентрации определяемого компонента удобнее использовать прямоугольные участки кинетических кривых. Характер кинетических кривых, а следовательно, и использование дифференциального или интегрального вариантов кинетических методов анализа определяется типом индикаторной реакции, ее механизмом. В настоящее время наиболее распространенными являются три основных способа определения неизвестной концентрации по данным кинетических измерений. Это способы тангенсов, фиксированного времени, фиксированной концентрации. Рассмотрим их применительно к дифференциальному варианту кинетического метода анализа. Способ тангенсов основан на определении тангенса угла наклона кинетических кривых tg при известных концентрациях определяемого вещества. При этом tg характеризует скорость индикаторной реакции и зависит от концентрации определяемого вещества. Градуировочный график строят в координатах: концентрация определяемого соединения — tg (рис. 1, а). Способ фиксированного времени. При определенном, строго фиксированном интервале времени протекания реакции, измеряют концентрацию индикаторного вещества в пробах с известными концентрациями определяемого компонента. Градуировочный график строят в координатах концентрация определяемого вещества — концентрация индикаторного вещества при фиксированном времени протекания реакции tфикс. (рис. 1, б). Часто при работе этим методом индикаторную реакцию останавливают при tфикс.. Путем резкого охлаждения, изменения кислотности раствора, добавления ингибиторов. Способ фиксированной концентрации. В отдельных пробах с известными концентрациями определяемого вещества проводят индикаторную реакцию до строго определенной (фиксированной) концентрации индикаторного вещества хфикс. и измеряют время достижения этой концентрации. Градуировочный график строят в координатах: концентрация определяемого компонента — величина, обратная времени достижения хфикс. (рис.1, в). В интегральном варианте все способы определения неизвестной концентрации аналогичны, лишь между временем реакции и концентрацией индикаторного вещества существует более сложная функциональная зависимость. В этом случае находят функции концентрации индикаторного вещества, линейно изменяющиеся во времени (логарифмическая, обратная и т. д.).
Рис. 1. Способы определения неизвестной концентрации по данным кинетических измерений: а — тангенсов; б фиксированного времени; в — фиксированной концентрации
(х — концентрации индикаторного вещества, t — время, с3 > c2
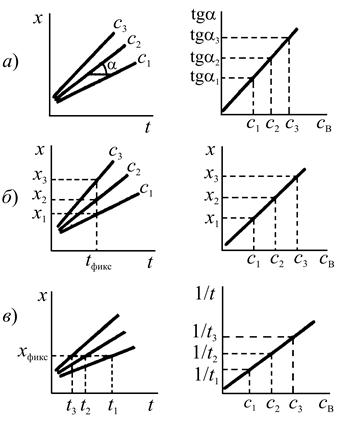
> c1 — концентрации определяемого соединения Каталиметрическое титрование процесс титрования в присутствии катализатора, в котором точку конца титрования определяют по резкому увеличению или уменьшению скорости реакции. С целью автоматизации каталиметрического метода анализа скорость реакции часто измеряют в открытых системах. Открытой называют систему, в которую по мере протекания реакции вводят реагенты и/или из которой отводят продукты реакции. В ходе реакции растворы подаются в реакционную камеру с постоянной или регулируемой скоростью. Разработаны разные варианты открытых систем: на основе проточных методов и «стат»-методов. Проточные методы. К ним относится метод непрерывной струи, основанный на смешении реагентов в струе и предложенный для быстро протекающих реакций с периодом полупревращения t1/2 = 0,01–10 с. Другой вариант проточного метода применяют для измерения скоростей сравнительно медленно протекающих реакций с t1/2 = 1–10 мин. В этом случае проточная ячейка одновременно является и смесительной камерой. Исходные реагенты индикаторной реакции и анализируемый раствор, содержащий катализатор с концентрацией скат, непрерывно подаются насосами в смесительную камеру вместимостью около 10 мл, продукты реакции и реагенты вытекают со скоростью 2–20 мл/мин. При каждом значении скат устанавливается постоянная концентрация индикаторного вещества и фиксируется постоянный сигнал, соответствующий скат. Смена раствора в кювете происходит за 1–2 мин, что определяет производительность анализатора 30 проб в час. Стат-метод предполагает введение реагентов со скоростью, равной скорости их расходования в реакции, так что концентрация индикаторного вещества остается постоянной. Скорость введения реагента регулируется автоматически. Воспроизводимость результатов кинетических измерений повышается при использовании метода одновременного компарирования. В анализируемый раствор и растворы шкалы стандартов одновременно с помощью стартовой пипетки вводят реагент, инициирующий протекание каталитической реакции. Через определенный промежуток времени сравнивают аналитические сигналы анализируемого раствора и шкалы стандартов и оценивают содержание определяемого вещества. Метод не требует термостатирования. Для учета влияния примесей на скорость реакции применяют метод добавок. Скорость реакции измеряют в равных аликвотных частях анализируемого раствора без добавки и в присутствии определенных добавок катализатора. Метод добавок дает правильные результаты, если в растворе отсутствуют посторонние примеси, обладающие каталитическим действием на индикаторную реакцию. Скорость реакции можно определять по времени внезапного появления окраски раствора в реакциях Ландольта. Реакции Ландольта — это медленные химические реакции, в которых образование окрашенного продукта реакции задерживается подходящим реагентом, специально добавленным для этой цели. Например, при окислении бромида персульфатом, катализируемом ионами меди(II), образующийся бром окисляет аскорбиновую кислоту и не взаимодействует с индикатором N,N-диметил-п-фенилендиамином. Когда практически вся аскорбиновая кислота окислится, появляется окраска индикатора. Метод, основанный на эффекте Ландольта, в ряде случаев обеспечивает более высокую воспроизводимость результатов анализа, чем обычный метод фиксированной концентрации, разновидностью которого он является. Концентрацию катализатора можно определять по длительности индукционного периода tинд., по истечении которого скорость реакции становится заметной (рис. 2). Этот способ является разновидностью метода фиксированной концентрации. Индукционный период наблюдается не только в реакциях Ландольта, но и в автокаталитических реакциях, а также в реакциях, когда в начальный период изменяется соотношение форм катализатора. Длительность индукционного периода связана с концентрацией катализатора зависимостью
Рис. 2. Кинетические кривые окисления KI (2 10–4 М) пероксидом водорода (2,4 * 10–3 М), катализируемого Ti(IV), в присутствии крахмала За изменением концентрации индикаторного вещества во времени можно наблюдать любым методом, и при построении кинетических кривых вместо концентрации образующегося продукта использовать любую, пропорциональную ей величину — оптическую плотность, силу тока, потенциал системы и т. д. Чаще всего для наблюдения за скоростью индикаторной реакции используют спектрофотометрические и люминесцентные, реже электрохимические, термометрические и титриметрические методы. Некаталитические методы не отличаются высокой чувствительностью (она определяется, как правило, методом наблюдения за скоростью индикаторного процесса), но они селективны, часто позволяют определять в смеси близкие по свойствам вещества без их предварительного разделения. Эти методы применяют при анализе смесей органических соединений (спиртов, сахаров, аминов) и смесей таких близких по свойствам ионов металлов, как щелочно-земельные и редкоземельные элементы. Каталитические методы анализа отличаются высокой чувствительностью, которая для многих неорганических веществ сравнима с чувствительностью масс-спектральных и активационных методов анализа, а для органических — с наиболее чувствительными вариантами хроматографии. В отдельных случаях, например, для серебра, хрома, кобальта, каталитические методы — наиболее чувствительные из всех известных методов анализа. При этом преимуществом каталитических методов является сочетание высокой чувствительности с простотой аппаратурного оформления и методики проведения анализа. Среди каталитических методов высокую чувствительность и селективность имеют ферментативные методы, основанные на использовании реакций, катализируемых ферментами. Ферментативными методами определяют субстраты, сами ферменты и эффекторы ферментов (соединения, мешающие активности ферментов). Методы определения субстратов — веществ, на которые действуют ферменты — высокоселективны и даже специфичны, что позволяет определять субстраты непосредственно в матрице сложных объектов (кровь, биомассы и биожидкости, многокомпонентные технологические растворы). Чувствительность определения при этом обусловлена методом, выбранным для контроля за скоростью процессов. Часто в этих случаях используют ферментные электроды. Методы определения эффекторов ферментов высокочувствительны, но не всегда селективны. В кинетических методах наиболее часто используют метод тангенсов как наиболее точный (использует большое число экспериментальных данных) и универсальный (применим для реакций с индукционным периодом). Реже применяют способ фиксированного времени и способ фиксированной концентрации, хотя эти способы более просты и менее трудоемки. Способ фиксированной концентрации используют обычно при автоматизации контроля, способ фиксированного времени — при проведении серийных анализов. Метод каталиметрического титрования применяют для определения с повышенной точностью микросодержания ионов металлов или органических соединений, образующих с ионом металла устойчивые, каталитически неактивные комплексы, и следов органических веществ в неорганических солях особой чистоты. При титровании органического соединения избыток титранта (иона металла-катализатора) уже в концентрации 10–8–10–6 М вызовет протекание каталитической реакции и тем самым определит конечную точку титрования . Основное применение каталитических методов в анализе реактивов и веществ особой чистоты определение микроконцентраций переходных металлов. Именно каталитические методы позволяют определить ионы элементов, содержание которых часто лимитируется в технических условиях на вещества особой чистоты табл. 1. Чистота цитирования примесей* в веществах особой чистоты [16] и пределы обнаружения их каталиметрическими методами
Чаще всего лимитируется содержание железа и меди (допустимый уровень — 10–4–10–6%). Другие примеси определяют реже, но для некоторых веществ необходим контроль их содержания на уровне 10–7%, который не обеспечивается традиционными методами эмиссионной спектроскопии и спектрофотометрии. Особенно успешно каталитические методы применяются для определения Co, Mn, V, Mo, W, Nb, Ta. Кроме того, при анализе веществ особой чистоты каталитические методы позволяют определять отдельные анионы, органические соединения в неорганических солях, отклонения от стехиометрии в составе соединений. Нижняя граница концентраций, определяемых с применением каталитических методов, для большинства элементов составляет 10–8–10–9 М (10–3–10–4 мкг/мл), т. е. на 1–2 порядка ниже, чем в методах спектрофотометрии, полярографии и пламенного варианта атомной абсорбции. Скорость конкретной индикаторной реакции зависит от многих параметров: температуры, ионной силы раствора, концентрации мешающих компонентов, наличия ингибиторов и активаторов. Все эти параметры в кинетических методах анализа необходимо строго контрфолировать и поддерживать постоянными для получения надежной аналитической информации. Каталитическая реакция является
результатом протекания ряда элементарных процессов, поэтому влияние температуры
на скорость процесса не всегда легко интерпретировать. Для практической оценки
влияния температуры на скорость реакции используют температурный коэффициент
Кинетическим определениям могут мешать посторонние ионы и соединения: · обладающие каталитической активностью; ·
снижающие скорость
каталитической реакции или изменяющие направление ее отдельных стадий. Мешающее
влияние оказывают окислители и восстановители, взаимодействующие с реагентами
индикаторной реакции и катализатором: Mn(VII), Cr(VI), I2, Избирательность индикаторных реакций характеризуется максимально допустимым содержанием посторонних ионов, при которых относительная погрешность определения анализируемого вещества не превышает заданного значения, например, 0,1. Кислотность раствора влияет на
избирательность определения, если в лимитирующей стадии индикаторной реакции
участвует ион водорода, а также в случае образования в результате гидролиза при
разных значениях рН гидроксокомплексов, обладающих различной каталитической
активностью. Так, каталитически активные комплексы Ti образуются при рН = 3,8,
а максимальное содержание каталитически активных Для устранения мешающего влияния посторонних компонентов применяют традиционные для аналитической химии приемы отделения или маскирования мешающих примесей. Кинетические методы (в том числе и ферментативные) при условии строгого соблюдения условий проведения анализа не уступают другим методам по точности, достаточно экспрессны, легко поддаются автоматизации. В практике аналитической химии эти методы применяют: · при анализе смесей близких по свойствам органических соединений (некаталитический вариант); · при определении микроколичеств ионов 3d-элементов, группы платиновых металлов, ряда анионов (I , Cl , Br ) и органических веществ в природных и промышленных объектах; · при определении присутствия токсичных и лекарственных препаратов, физиологически активных соединений в биологических объектах и объектах окружающей среды (ферментативные методы). ГЛАВА 2. НЕКОТОРЫЕ ОСНОВНЫЕ СВЕДЕНИЯ О ФЕРМЕНТАХ Ферменты - биологические катализаторы белковой природы, ускоряющие химические реакции в живых организмах и вне их. Ферменты обладают уникальными свойствами, которые выделяют их на фоне обычных органических катализаторов гомогенного типа. Прежде всего это необычайно высокая каталитическая активность. Так, добавка незначительной концентрации фермента (10- 9-10- 7 М) ускоряет превращение субстрата в 108-1012 раз. Другое, не менее важное свойство ферментов - специфичность (избирательность) их действия в отношении структуры субстрата, типа реакции и условий ее проведения. Специфичность определяется способностью фермента превращать только данный тип субстратов в определенных реакциях и условиях. Эти свойства ферментов обусловлены сложной структурой макромолекул белка и весьма сложным механизмом их действия. Превращение субстрата происходит на активном центре фермента. Для многих ферментов, состоящих из субъединиц, характерно наличие регуляторного участка, который взаимодействует с веществами, влияющими на активность фермента, - активаторами, ингибиторами или инактиваторами (рис. 4). Важным свойством ферментов, которое необходимо учитывать при их практическом использовании, является стабильность, то есть способность сохранять каталитическую активность. При хранении и особенно в ходе ферментативной реакции фермент может частично или полностью терять свою каталитическую активность, другими словами, инактивироваться. Одним из эффективных способов стабилизации ферментов является их иммобилизация - перевод в водонерастворимое состояние путем связывания с носителем или модифицирование водорастворимыми полимерами с полным или частичным сохранением ферментами каталитической активности. Повышение стабильности и возможность многократного использования иммобилизованных ферментов значительно повышают экономичность ферментативных методов анализа. 2. 1. Возможности ферментативных методов анализа Пределы обнаружения веществ, определяемых ферментативными методами (субстратов, активаторов, обратимых и необратимых ингибиторов и инактиваторов ферментов) зависят не только от каталитической активности фермента, но и от других кинетических характеристик используемой индикаторной реакции. Фермент катализирует реакции, в которых участвуют, как правило, один или два субстрата. Простейшая односубстратная реакция описывается обычно схемой Михаэлиса-Ментен E + S ES P + E, где Е - фермент, S - субстрат, ES - промежуточный комплекс фермента с субстратом (комплекс Михаэлиса-Ментен), Р - продукт. Концентрация субстрата [S]0 будет пропорциональна u0 только при условии, когда [S]0 ! Km . Тогда [S]0 = u0Km / kкат[E] Следовательно, верхняя граница определяемых концентраций субстрата ограничена величиной константы Михаэлиса-Ментен (Km). Нижняя граница определяемых концентраций субстрата определяется той величиной u0 , которая может быть зафиксирована с помощью используемого для наблюдения инструментального метода, причем величина u0 тем выше для одного и того же значения [S]0 , чем выше kкат и [E]. Таким образом, использование высокоактивного фермента (большое значение kкат) и повышение его концентрации в реакционной смеси могут существенно снизить предел обнаружения субстрата. Несколько другие кинетические закономерности следует учитывать при определении тех веществ, которые являются эффекторами ферментов. Для
контроля начальной скорости ферментативного процесса или концентрации продукта
реакции могут быть использованы практически любые доступные исследователю
физико-химические методы. Наиболее часто применяют фотометрические методы
индикации. При этом в качестве индикаторных часто используют, например,
катализируемые пероксидазой и другими оксидазами (ферментами, катализирующими
окислительно-восстановительные реакции), а также гидролазами (катализирующими
реакции гидролиза) реакции образования окрашенных продуктов. Исключительно
высокой чувствительностью отличаются хемилюминесцентные методы, позволяющие
контролировать скорость ферментативных реакций с участием, например,
люциферазы. Различные электрохимические методы (потенциометрия, амперометрия)
наиболее удобны для контроля скорости реакций, протекающих с поглощением или
выделением протонов, а также окислительно-восстановительных процессов,
сопровождающихся поглощением кислорода, образованием Н К настоящему времени разработано большое число высокочувствительных и селективных ферментативных методов определения субстратов и эффекторов ферментов: неорганических (ионов металлов, анионов) и органических (N-, S-, P-, O-содержащих) соединений. При этом можно выделить ферменты, позволяющие определять целый класс соединений, либо часть этого класса, либо индивидуальное соединение. Так, с помощью алкогольдегидрогеназы можно определять спирты (субстраты этого фермента), алкогольоксидазы - первичные спирты, арилалкогольоксидазы - ароматические первичные спирты. Следует отметить, что методы определения эффекторов, хотя и менее селективны, часто более чувствительны, чем методы определения субстратов. Так, пределы обнаружения многих органических веществ - субстратов ферментов лежат в интервале 10- 6-10- 4 М, а органических эффекторов - 10-11-10- 8 М. Кроме того, число эффекторов ферментов значительно больше, чем число их субстратов, что расширяет возможности ферментативных методов анализа. 2. 2. Определение органических соединений Ферментативные методы определения органических соединений успешно разрабатываются и применяются в клинических и биохимических лабораториях. Именно применение ферментов дает возможность селективно определять в крови, моче, тканях и других биологических объектах малые количества таких метаболитов и физиологически активных веществ, как мочевина, мочевая кислота, аминокислоты, сахара (в частности, глюкоза), спирты, липиды, холестерин, нуклеотиды, антибиотики. Ферментативное определение мочевины основано на реакции ее гидролиза, катализируемой ферментом уреазой. Образующиеся продукты гидролиза - ионы и можно определять электрохимически, фотометрически или флуориметрически. Групповое определение аминокислот основано на использовании таких ферментов, как L-амино- или D-аминооксидаза, которые катализируют окисление аминокислоты кислородом воздуха до кетокислоты, пероксида водорода и аммиака. Последний далее определяют электрохимически с помощью NH3-чувствительного газового электрода. Специфическое определение отдельных аминокислот возможно при применении декарбоксилаз, дегидрогеназ, лиаз, трансфераз. Для определения глюкозы используют несколько специфических ферментативных реакций, например: 1) катализируемое глюкозооксидазой окисление ее кислородом воздуха (или другими окислителями) до глюконовой кислоты и пероксида водорода; 2) ее взаимодействие с АТФ с образованием глюкозо-6-фосфата в присутствии гексокиназы. При использовании первой реакции определение глюкозы проводят либо наблюдая за уменьшением количества кислорода в растворе с помощью О2-чувствительного электрода Кларка, либо измеряя рН раствора, который изменяется вследствие образования глюконовой кислоты, либо определяя количество образовавшегося пероксида водорода. Ферментативные методы определения сахарозы и других дисахаридов основаны на использовании специфических ферментов (инвертазы, лактазы), превращающих дисахариды в моносахариды, одним из которых является глюкоза. Далее глюкозу определяют с помощью описанного выше метода. Ферментативное определение этанола основано на использовании одного из двух ферментов: алкогольоксидазы, катализирующей окисление этанола кислородом воздуха до ацетальдегида и Н2О2 , или алкогольдегидрогеназы, в присутствии которой спирт в реакции с НАД (никотинамидадениндинуклеотидом) превращается в НАД " Н2 . Определение различных токсичных соединений, таких, как фенолы, ароматические амины, фосфор-, сероорганические соединения, является важной задачей аналитической химии и в первую очередь связано с проблемами контроля и охраны окружающей среды. Некоторые токсичные соединения являются субстратами специфических ферментов, что позволяет значительно упростить подготовку проб сложных по составу объектов анализа. Например, определять мочевину в воде плавательных бассейнов, почве позволяет использование уреазы, для которой мочевина является высокоспецифическим субстратом. Однако в большинстве предложенных методов для определения токсичных соединений используют их ингибирующее действие на ферменты. Например, фосфорсодержащие пестициды определяют по ингибированию холинэстеразы или карбоксилэстеразы; серосодержащие ингибиторы, различные фенолы - с помощью пероксидазы; по ингибированию свечения бактериальной люциферазы определяют инсектициды: ДДТ, пентахлорфенол. Для определения фунгицидов и инсектицидов в плодах, ягодах, листьях применяют папаин, ацетилхолинэстеразу. 2. 3. Определение неорганических соединений Ферментативные методы успешно применяют для чувствительного и селективного определения ионов металлов, неорганических анионов, пероксида водорода, кислорода, растворенного в воде. Многие ионы металлов (например, Ag, Cu, Hg, Zn, Bi, Cd) можно определять с применением ферментов в количествах, недоступных определению с помощью большинства физико-химических методов анализа. Так, с применением щелочной фосфатазы разработан метод определения нанограммовых количеств бериллия. По ингибирующему действию на алкогольдегидрогеназу возможно определять ионы серебра в концентрации 10 пг/мл. Предложенные методы определения ртути по ингибирующему действию на пероксидазу и уреазу являются одними из самых чувствительных методов. Важно отметить, что приведенные методы являются не только высокочувствительными, но и селективными. Обычно же методы, основанные на ингибирующем или активирующем действии ионов металлов на фермент, не отличаются высокой селективностью. Однако избирательность методов можно повысить при использовании обычных для аналитической химии приемов маскирования. Наиболее чувствительными и селективными являются ферментативные методы определения ионов металлов по реактивации апофермента (белковой части фермента). Так, для определения цинка и кальция использована реактивация апофермента, полученного удалением ионов цинка из щелочной фосфатазы. Чувствительный и селективный метод определения меди основан на реактивации апофермента, полученного диализом иммобилизованной полифенолоксидазы. Для определения анионов, таких, как CN-, чаще всего используют ферментные электроды, позволяющие проводить экспрессный анализ сложных промышленных и биологических объектов. Как правило, в ферментных электродах применяют иммобилизованные ферменты. Чаще всего анионы являются субстратами в тех ферментативных реакциях, которые положены в основу методов их определения. Пределы обнаружения анионов при использовании ферментных электродов обычно выше 10 мкМ. Следует особо выделить высокочувствительные методы определения таких физиологически активных анионов, как CN-, I-, S2 -, F -, основанные на их реактивирующем действии на инвертазу, ингибированную ртутью или серебром (с пределами обнаружения 0,2-10 нг/мл), а также на ингибирующем действии на пероксидазу и кислую фосфатазу. Определение других неорганических соединений - аммиака, кислорода, диоксида серы, пероксида водорода основано на том, что они являются субстратами многих ферментов и, следовательно, могут быть определены с их помощью. Расширению применения ферментов в аналитической химии способствуют дальнейшее развитие энзимологии, разработка новых способов стабилизации ферментов, выделение и исследование новых ферментов. ГЛАВА 3. ПРИМЕРЫ ИСПОЛЬЗОВАНИЯ КИНЕТИЧЕСКИХ МЕТОДОВ В АНАЛИЗЕ ОБЪЕКТОВ ОКРУЖАЮЩЕЙ СРЕДЫ Исследование влияния неблагородных металлов на кинетическое определение родия Развитие аналитической химии малых содержаний платиновых металлов может быть связано с совершенствованием кинетических методов их анализа. Родий - один из элементов группы платиновых металлов, число индикаторных реакций для определения которого кинетическим методом невелико. Наиболее избирательной и чувствительной (2 10-4 мкг/мл Rh) является реакция окисления меди (II) периодат-ионом с образованием окрашенного периодатного комплекса меди (III) в щелочной среде (pH=8,3 - 8,5, =400 нм), которая лежит в основе кинетического определения родия методом тангенсов. Определению родия не мешают 20-кратный избыток платины, палладия, иридия и золота, что вполне устраивает аналитиков, если речь идет об анализе технологических растворов. Однако, такие растворы содержат большие количества железа (1:13000), калия (1:400) и натрия (1:26000). Имеющиеся литературные данные свидетельствуют, что железо (III) уже в количествах в 25 раз превышающих содержание родия искажает результаты анализа, а данные о мешающем влиянии натрия и калия вообще отсутствуют. В данной работе исследовались различные методы устранения мешающего влияния железа (III) на указанную выше кинетическую реакцию и исследовалось мешающееся влияние натрия и калия. Нами установлено, что описанные в литературе методы маскирования железа (III) фосфат- и фторид- ионами, лимонной, винной кислотами неэффективно в оптимальных условиях кинетического определения родия. Следовательно, для анализа таких растворов необходима предварительная стадия переведения железа в малорастворимое соединение с целью отделения от родия, причем на этой стадии весь родий должен оставаться в растворе. При осаждении гидроксида железа 2 М раствором гидроксида натрия 86,7 % родия увлекается в осадок, а при осаждении 25%-ным раствором аммиака, в осадок переходит 76,7 % родия и лишь использование еще более слабого основания (нитрата натрия) позволяет в достаточной мере достигнуть переведения железа в осадок без осаждения родия. СПЕКТРОФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ПЕРСУЛЬФАТА АММОНИЯ С ИСПОЛЬЗОВАНИЕМ КИНЕТИЧЕСКОГО МЕТОДА Как известно, персульфат аммония используется в практике аналитической химии, в том числе при анализе различных цветных сплавов, стали и чугунов. Кроме того, важен вопрос аналитического контроля продуктов получения персульфата аммония, производство которого стало необходимым в последнее время по следующим причинам. В технологии аффинажа платиновых металлов для очистки палладия от платины требуется сильный окислитель, как им и является персульфат аммония в аммиачной и нейтральной средах. Товарная его форма в виде соли или раствора обеспечивает удобство применения его по сравнению с другими окислителями. Несмотря на широкий круг потребителей в других отраслях промышленности выпуск персульфата аммония в России прекращён и поставки его обеспечивается только импортом. Поэтому остро стоит задача разработки технологии производства и анализа данного реагента на месте его потребления. Для определения малых количеств (10-60 мкг) персульфат-иона использовали кинетический метод со спектрофотометрическим контролем иодометрической реакции. Светопоглощение измеряли при постоянном значении рН, равном 6.85 (регулируемый показатель кислотности реакции) и при длине волны 364 нм. Кинетические кривые зависимости .оптическая плотность-время обрабатывали методом тангенсов. Содержание персульфата аммония определяли методом градуировочного графика (зависимость tg- концентрация определяемого вещества). Для определения персульфата аммония в интервале 50-260 мкг использовали описанный выше метод, но следует заметить, что нарастание оптической плотности, связанное с выделением йода, происходит очень быстро: в течение 3 минут при концентрации 0.2 мг. Поэтому при концентрациях 0.25-0.75 мг вещества в описанной выше методике использовали тиосульфат натрия в качестве замедлителя. Относительная погрешность определения не превышала 3%. КИНЕТИЧЕСКИЕ ЗАКОНОМЕРНОСТИ НЕФТЯНОЙ ИНТОКСИКАЦИИ СЕГОЛЕТОК РУССКОГО ОСЕТРА Для оценки и прогноза изменений состояния природных экосистем широко используются непрерывные или регулярные измерения их физических, химических и биологических параметров, лежащие в основе экологического мониторинга. Между тем экосистемы достаточно долго сохраняют "следы" прошлого, с помощью которого можно не только реконструировать пройденный ими путь развития, но вкупе с оценкой текущего состояния также составить прогноз того, что их ожидает в будущем. Такие исследования мы называем экологической диагностикой, поскольку в отличие от экологического мониторинга они могут носить единовременный характер. Наибольших успехов экологическая диагностика достигла в области биохимии и физиологии последствия перенесенных организмами стрессов порой ощущаются в течение всей их жизни. По нашему мнению, широкие возможности для развития экологической диагностики открывает также биотестирование, если оно сочетается с изучением кинетических закономерностей воздействия внешних факторов на биологические системы. При биотестировании природных вод обычно встают два вопроса: токсична ли данная природная вода, а если токсична, то какова степень ее токсичности. Наличие токсичности устанавливают с помощью набора тест-организмов, включающего, как правило, представителей основных трофических уровней экосистемы - водоросли, зоопланктон, рыбы. Главное требование при их выборе - это высокая чувствительность к токсичному веществу. Значительные сложности возникают при оценке степени токсичности природных вод. Существующие методы ее оценки можно условно разделить на три группы. Первая группа предусматривает разработку предельно допустимой концентрации (ПДК) токсичного вещества путем определения таких параметров тест-объектов, как летальная или эффективная концентрация для 50% организмов (ЛК50 или ЭК50), острое и хроническое действие токсичного вещества (ОТД или ХТД) на "выживаемость", "плодовитость", "изменение роста" и т.д., ориентировочно безопасного уровня содержания токсичного вещества (ОБУВ) или его максимальной недействующей концентрации (МНК) и других аналогичных параметров. При всей привлекательности нормирования природных вод с помощью ПДК или подобных ему показателей, оно, строго говоря, антиэкологично. Во-первых, такое нормирование признает существование нижнего безопасного порога концентрации токсиканта в объектах биосферы, что во многих случаях, например, при оценке воздействия канцерогенов, далеко не бесспорно, и, во-вторых, в природных водах присутствуют сотни и тысячи химических веществ, которые проявляют друг к другу ингибирующее, аддитивное и синергическое действие, а многие безвредные вещества в определенных условиях становятся токсичными. Принцип ПДК, равно как ОБУВ или МНК, может быть использован для лимитирования сброса токсичного вещества в водоем, но не для отображения экологического состояния самого водоема. Второй способ основан на разработке шкал токсичности, например, с помощью формулы Хабера E = C·t, (1) где E - эффект; C - концентрация токсичного вещества; t - время действия. Критерий Хабера лишен характерных для ПДК недостатков, однако он позволяет выразить степень токсичности только качественно, как гипер-, остро-, умеренно-, слаботоксичная и не токсичная. К третьей группе можно отнести количественные способы выражения степени токсичности природных вод. Наибольшее распространение среди них получил способ оценки токсичности в баллах с помощью так называемой формулы "суммарной токсичности" [1]: tХ = 1T(i)/(tT(i)·n), (2) где tХ - "суммарная токсичность"; 1T(i) - условный балл класса реакций тест-организмов; tT(i)- время проявления реакции тест-организма; n - число тест-организмов, у которых отмечена соответствующая реакция. Согласно (2), чем больше значение tХ, тем выше балл токсичности, и по этому критерию уровень токсичности природной воды изменяется от нетоксичной (балл токсичности = 0) до чрезвычайно токсичной (балл токсичности = 500). Следовательно, критерий tХ, также как и ПДК, "признает" существование нижнего порога действия токсичного вещества. Бальную систему используют также при ранжировании токсичности по кратности разбавления (при нормировании сброса сточных вод) и при оценке экологического статуса водоема в терминах "олиго-политоксичность" (при классификации вод по сапробности). Особо следует сказать о прогностических возможностях существующих критериев оценки токсичности природных вод. Критерии (1) и (2), фиксируя физиолого-морфологические реакции тест-организма, с той или иной степени достоверности отражают состояние природных вод в данный момент времени, но они не могут быть использованы для ранней диагностики токсического воздействия того или иного вещества. Между тем, в условиях все возрастающего антропогенного воздействия на окружающую среду, вопросы раннего прогнозирования устойчивости природных экосистем становятся приоритетными. Иными словами, речь идет об оценке изменений, происходящих в экосистеме до проявления морфологических, физиологических, популяционных и других отклонений от нормы и их использования для предсказания возможных путей развития экосистемы. Это обстоятельство вынуждает перенести акцент мониторинговых исследований природных вод на изучение биохимических процессов, происходящих в тест-организма при воздействии токсичного вещества. Надо заметить, что проблемам биохимии водных организмов в последнее время уделяется огромное внимание, однако при попытке использовать их в прогностических целях возникают существенные, предопределенные сложностью и многообразием биохимических процессов, трудности. Предлагается иной подход к оценке токсичности природных вод. Он основан на: а) абстрагировании от механизма биохимического воздействия загрязняющего вещества на тест-организм и б) использовании для описания реакции тест-организма на загрязняющее вещество представлений формальной химической кинетики, основанных на принципе лимитирующей (или скорость определяющей) стадии сложных процессов. Действительно, любой отклик тест-объекта на токсичное вещество можно представить, как результатом химической реакции между "активными центрами" тест-организма и токсичным веществом. Тогда формально кинетика отклика тест-организма на токсичное вещество можно записать в виде: ±dZ/dt = k·C1·C2 = k·a·C1·C3, (4) где Z - положительный (+) или отрицательный (-) отклик тест-организма; t - время; C1 - концентрация активных центров тест-организма; C2 - концентрация токсиканта в фазе тест-организма; C3 - концентрация токсиканта в водной фазе; a - коэффициент пропорциональности между С2 и С3; k - константа скорости реакции отклика тест-организма. Ясно, что в качестве количественного критерия степени токсичности природных вод здесь выступает параметр k. Распространение представлений формальной химической кинетики на биотестирование, проведя при этом некоторую аналогию между сорбционными свойствами полимерных и биологических мембран, позволяет предвидеть два крайних случая: С1=k1C2 и C2>>C1. Тогда уравнение (4) можно переписать в виде ±dZ/dt = k1·C22, (5) или в виде ±dZ/dt = k2·C1, (6) где k1 = k·С1; k2 = k·C2 = k·a·C3. Из последних выражений следует, что в зависимости от соотношения концентраций активных центров тест-организма и токсичного вещества, реакция отклика тест-организма может быть как бимолекулярной, так и мономолекулярной. С целью апробирования предлагаемого подхода для мониторинговых исследований изучалось воздействие нефтяного загрязнения на сеголетки русского осетра. В качестве тест-реакции на нефтяные углеводороды (НУ) было использовано изменение концентрации суммарных сульфгидрильных групп в тканях сеголеток. Для этого по 12 экземпляров сеголетки русского осетра длиной тела 6-8 см помещали в аквариумы объемом 20 л с различным содержанием сырой нефти. Предварительно нефть эмульгировали перемешиванием механической мешалкой в течение 10 минут при комнатной температуре. Смена раствора проводили через каждые 3 суток. По истечении заданного времени рыбы извлекали из аквариума и подвергались биохимическому анализу. Параллельные измерения проводили на контрольных рыбах, содержащих в аквариуме без нефти. В процессе опыта рыб кормили живым трубочником. Сульфгидрильные группы в тканях сеголеток, солюбилизированных додецилсульфатом натрия, определяли методом амперометрического титрования, в качестве титранта использовали нитрат серебра. Выбор в качестве тест-реакции изменение содержания SH-групп в тканях сеголеток обусловлен следующим. Сульфгидрильные группы выделяются среди других функциональных групп рыб высокой реакционной способностью и многообразием химических реакций, в которые они вступают - алкилирование, ацилирование, окисление, тиолдисульфидный обмен, образование меркаптидов, полумеркаптилий, меркапталов, водородных связей и комплексов с переносом заряда. Во многих реакциях SH-группы принимают участие в форме меркаптидного иона, обладающего высокой нуклеоофильной способностью, они легко окисляются молекулярным кислородом, его радикалами, перекисью водорода. Среди процессов, протекающих в организме с участием SH-групп белковой и небелковой природы, следует отметить ферментативные реакции - в настоящее время насчитывается около 100 ферментов, в активности которых принимают участие SH-групп. SH-групп играют важную роль в ряде физиологических и биохимических процессов - в нервной деятельности, мышечном сокращении, делении клеток, регуляции проницаемости мембран митохондрий, окислительном декарбоксилировании a-кетакислот, окислительном фосфолировании, фотосинтезе, механизме радиационных поражений и при действии токсичных веществ. Из сказанного следует, что концентрация SH-групп должна тонко реагировать на присутствие в природной воде токсичного вещества.
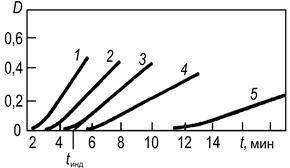
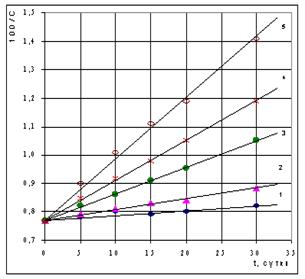
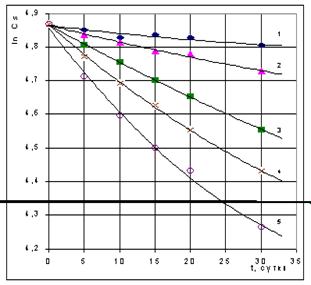
Рис. 1. Изменение концентрации сульфгидрильных групп в теле личинок русского осетра при их экспозиции в воде, содержащей O.05 (1), 0.25 (2), 0.50 (3), 0.75 (4) и 1.00 (5) мг/л нефти. Точки - экспериментальные данные, сплошные линии - расчет по уравнению (14). Полученные экспериментальные данные приведены на рис. 1. Они показывают, что выдерживание рыб в воде, содержащей нефть, приводит к уменьшению концентрации SH-групп белков и низкомолекулярных тиоловых соединений сеголеток русского осетра. Для краткости назовем этот процесс дезактивацией SH-групп, и применительно к нему перепишем уравнения (5) и (6) в виде: -dCSH/dt = k1C2SH, (7) -dCSH/dt = k2CSH. (8) Решение последних двух уравнений при начальных условиях, t = 0, CSH = C0SH, дает соответственно (9) и (10): CSH = C0SH/(1+k1tC0SH), (9) CSH = C0SH[1-exp(-k2t)], (10) где C0SH и CSH - концентрация сульфгидрильных групп в теле сеголеток до и после воздействия НУ в течение времени t. Построение анаморфоз уравнений (9) и (10) показала (рис. 2), что кинетика процесса дезактивации сульфгидрильных групп сеголеток лучше подчиняется уравнению реакции второго порядка, чем уравнению реакции первого порядка. Найденные по тангенсу угла наклона прямых 1/CSH=f(t) значения величин k1 приведены в табл. 1. Таблица 1. Константы скорости дезактивации SH-групп сеголеток русского осетра при их экспозиции в воде, содержащей нефть
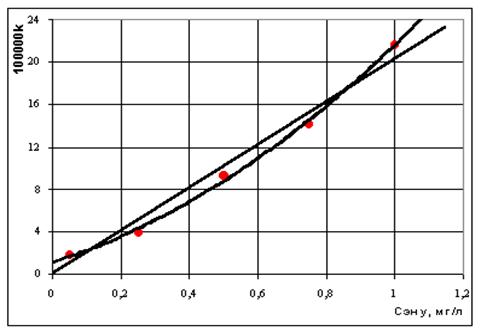
Рис. 2. К проверке выполнимости уравнений (9) и (10) для описания кинетики изменения концентрации SH-групп в теле сеголеток русского осетра. Обозначения, как на рис. 1. Полученные данные не позволяют однозначно подтвердить или однозначно опровергнуть вопрос о пороге токсического воздействия нефти на гидробионты. В пределах точности эксперимента зависимость константы скорости дезактивации SH-групп от концентрации нефти в воде (Сну) может носить либо линейный k1=20,23·Cну, (11) либо нелинейный k1=1,30+9,28·Cну+11,04·С2ну (12) характер (рис. 3). Очевидно, при оценке токсического воздействия нефтяных углеводородов на гидробионты необходимо исходить не из общего их содержания в воде, а из концентрации растворенных (активных) форм. Однако на практике не будет большой ошибкой, если исходить из предположения о беспороговом влиянии нефтяного загрязнения и принять, что k1 = b·Cну, (13) где b - коэффициент пропорциональности; Сну - содержание нефтяных углеводородов в воде, мг/л.
Рис. 3. Связь между константой скорости реакции дезактивации SH-групп сеголеток русского осетра и содержания нефти в воде. Точки – экспериментальные данные, сплошные линии - расчет по уравнениям (11) и (12). Если это предположение верно, то, объединив уравнения (9) и (13), получим выражение CSH = C0SH/(1+b·Cну·C0SH·t), (14) которое позволяет прогнозировать воздействие нефтяных углеводородов на концентрацию SH-групп в тканях сеголеток при любых значениях нефтяного загрязнения и при любых длительностях экспозиции. О точности выполнимости уравнения (14) можно судить по данным рис. 1, где сопоставлены экспериментальные значения SH-групп в тканях сеголеток с теоретически рассчитанными кривыми. В заключение отметим, что уравнение (12) позволяет решить и обратную задачу. Например, зная концентрацию SH-групп в тканях рыб, обитающих в загрязненной нефтью природной воде, можно найти время их пребывания в такой воде, а зная концентрацию SH-групп в тканях рыб и их возраст можно оценить "интегральное биологическое воздействие" нефтяного загрязнения природных вод на гидробионты. Кинетический метод определения йода в пищевых продуктах и продовольственном сырье Среди каталитических методов получил применение рода-нидно-нитритный метод. Он основан на реакции окисления роданид-иона смесью нитрат- и нитрит-ионов, катализируемой йодид-ионами. Предел обнаружения - 0,5-1,0 мкг в 100 г продукта. Среди различных методов определения йода в кормах и растениях (кинетический роданидно-нитрит-ный, кинетический церий-мышьяковистый, фотометрический с бриллиантовым зеленым, объемный йод-крахмальный) наибольшую чувствительность и воспроизводимость результатов дают кинетические рода-нидно-нитритный и церий-мышьяковистый методы. Первый из них рекомендован также для нужд агрохимической службы. Наряду с этим описан простой метод количественного определения общего йода в пищевых продуктах, основанный на каталитической деструкции тиоционата нитритом в присутствии йодида и последующем фотометрическом определении при длине волны 450 нм. Предел определения метода -1 мкг йода в 100 г продукта, правильность - 90%, стандартное отклонение - 10%. Колориметрический метод определения йодидов был оптимизирован для рутинного анализа йода в пищевых продуктах, подвергнутых кулинарной обработке и содержащих большое количество соли. Метод основан на деструкции ферро-тиоцианатного комплекса нитритом, катализируемым йодидом. Предел обнаружения - 2,5 мкг/л, интервал линейности аналитического сигнала - 2,5-12 мкг/л, возврат йодида и йодата -100±10%. По сравнению с МС-ИСП-методом установлено небольшое расхождение результатов (не более 5%). ГЛАВА 4. ОБОРУДОВАНИЕ ДЛЯ КИНЕТИЧЕСКИХ МЕТОДОВ АНАЛИЗА ФЛЮОРАТ®-02-АБФФ-Т полуавтоматический фотометрический анализатор биожидкостей Общее описание Выпускается по лицензии Министерства Здравоохранения Российской Федерации серии М № 011373 и имеет регистрационный № 29/07020299/0182-00 Лицензия № 64/2003-0280-0309 от 11 июля 2003 года Анализатор Флюорат®-02-АБФФ-Т - первый отечественный полуавтоматический фотометр медицинского назначения для определения биохимических показателей в биопробах. Анализаторы представляют собой открытую систему, т.е. предоставляет пользователю возможность работать с наборами реагентов любых фирм изготовителей. Анализаторы позволяют работать со следующими типами биохимических реакций: А) Реакции по конечной точке это тип реакций, в которых измерение оптической плотности рабочего раствора проводится после полного завершения химической реакции. Б) Кинетические реакции – тип реакций, в которых измерение оптической плотности рабочего раствора проводится в ходе протекания химической реакции. Кинетические методы анализа являются более точными и правильными, чем методы по конечной точке. В настоящее время эти методы все более широко используются в медицинской практике в России. В тоже время для реализации кинетических методов анализа требует специализированного оборудования, наличие в котором термостатирования измерительной кюветы с точностью не хуже ± 0,2 Со и специальной программы расчета - обязательное условие. Традиционно используемые в российских медицинских учреждениях фотометры общего назначения отечественного производства не дают возможности пользователям работать кинетическими методами. Анализатор Флюорат®-02-АБФФ-Т представляет собой фотометр и обеспечивает измерение основных типов реакций, применяемых в медицинской лабораторной практике: · Фотометрия по конечной точке со стандартом и холостой пробой по реагенту. · Фотометрия по конечной точке, по коэффициенту пересчета (фактору) и холостой пробой по реагенту. · Фотометрия по конечной точке со стандартом и холостой пробой по образцу. · Фотометрия по конечной точке, по коэффициенту пересчета (фактору) и холостой пробой по образцу. · Фотометрия, кривая калибровки по стандартам (до семи) с холостой пробой по реагенту. · Фотометрия. Кинетика (две точки) со стандартом. · Фотометрия. Кинетика (две точки) со стандартам и холостой пробой по реагенту. · Фотометрия. Кинетика. Фактор. · Оптическая плотность. · Бихроматика. Конечная точка по стандарту с холостой пробой по реагенту. Отличительные особенности: · Работа в диалоговом режиме (вопрос - ответ) · Автоматическая смена светофильтров при выборе методики измерения · Автоматическая загрузка в оперативную память калибровочной зависимости (калибровки) при смене методики измерения · Мембранный насос и проточная микрокювета (для фотометрических методик) · Малый расход реагентов 400 мкл 1000 мкл на 1 анализ · Встроенный термостат измерительной кюветы. Температуры стабилизации 25, 30, 37 оС · Хранение в памяти 40 методик анализа · Автоматический анализ результата на попадание в диапазон нормы · Печать протокола выполнения анализов на принтере · Производительность выполнения анализов до 100 анализов в час методом по конечной точке НПФ АП «Люмэкс»® выпустила уже второе поколение полуавтоматических биохимических анализаторов - «Фотометр®-АБФ-100» Принцип метода Измерение оптической плотности и автоматический расчет концентрации определяемого компонента. Анализаторы предназначены для работы с унифицированными в медицине фотометрическими методиками анализа для определения: общего белка, альбумина, общего и прямого билирубина, холестерина, триглицеридов, мочевины, АлТ, АсТ, ЛДГ, a-амилазы, 17- КС, 17- ОКС, кислой и щелочной фосфатазы и т.п., макроэлементов и электролитов. Процедура работы Алгоритм выполнения анализов предполагает сначала проведение специфической химической реакции (или ее начала) вне прибора (в пробирке). Для выполнения анализа используются специальные наборы реагентов, а в качестве пробы - соответствующий биологический материал. Далее: перенос рабочего раствора в кюветное отделение анализатора, измерение оптической плотности (интенсивности люминесценции) рабочего раствора, расчет концентрации, анализ результата на попадание в диапазон нормы, вывод результата и необходимых комментариев на экран и принтер. Все этапы работы анализатора кроме первых двух выполняются автоматически. Диалоговый режим работы, использованный в анализаторе - это наиболее удобный для пользователя режим, позволяющий максимально упростить работу оператора. В случае работы в этом режиме оператор просто выбирает необходимый пункт из меню, предлагаемого анализатором, или непосредственно выполняет инструкцию, которая в виде текста отображается на дисплее. Перед началом измерений в анализатор с клавиатуры необходимо ввести программу для выполнения анализа, в соответствии с инструкцией к используемому набору реагентов. Этот процесс также выполняется в диалоговом режиме. Области применения Анализ компонентов биопроб в клинико-диагностических лабораториях лечебных, профилактических, научно-исследовательских медицинских учреждениях. Области нибольшей эффективности работы прибора: · Малые и средние клинико-диагностические лаборатории медицинских учреждений или как второй фотометр в крупных лабораториях. Рекомендуемый комплект поставки · Основной блок анализатора · Стандартная кварцевая кювета К10 или блок проточной кюветы · Инструкция по эксплуатации · Сетевой предохранитель ЛИТЕРАТУРА 1. Золотов Ю.А., Дорохова Е.Н., Фадеева В.И. и др. Основы аналитической химии. В 2 кн. Кн. 2. Методы химического анализа. / Под ред. Ю.А. Золотова. М.: Высш. шк., 1996. 2. Основы аналитической химии. Практическое руководство. / Под ред. Ю.А. Золотова. М.: Высш. шк., 2001. 3. Крейнгольд С.У. Каталиметрия в анализе реактивов и веществ особой чистоты. М.: Химия, 1983. 4. Марк Г., Рехниц Г. Кинетика в аналитической химии. / Пер. с англ. Под. ред. К.Б. Яцимирского. М.: Мир, 1972. 5. Яцимирский К.Б. Кинетические методы анализа. Изд. 2-е. М.: Химия. 1967. 6. Номенклатура кинетических методов анализа. (Рекомендации ИЮПАК, 1993). // Журн. аналит. химии. 1998. Т. 53, № 1. С. 110–126. 7. Мюллер Х., Отто М., Вернер Г. Каталитические методы в следовом анализе. / Пер. с нем. Под ред. К.Б. Яцимирского. М.: Мир, 1982. 8. Сычев А.Я. Окислительно-восстановительный катализ комплексами металлов. Кишинев: Штиинца, 1976. 9. Набиванец Б.И., Линник П.Н., Калабина Л.В. Кинетические методы анализа природных вод. Киев: Наукова думка, 1981. 10. Бончев П. Комплексообразование и каталитическая активность. / Пер. с болг. Под ред. К.Б. Яцимирского. М.: Мир, 1975. 11. Каталог химических реактивов и особо чистых химических веществ. М.: Химия, 1983. 12. Крейнгольд С.У., Божевольнов Е.А., Драпкина Д.А. // Журн. аналит. химии. 1967. Т. 22, № 3. С. 218. 13. Диксон М., Уэбб Э. Ферменты. М.: Мир, 1982. Т. 1. 389 с. 14. Фершт Э. Структура и механизм действия ферментов. М.: Мир, 1980. Т. 1. 432 с. 15. Клячко Н.Л. Ферменты - биологические катализаторы: Основные принципы действия // Соросовский Образовательный Журнал. 1997. № 3. С. 58-63. 16. Келети Т. Основы ферментативной кинетики. М.: Мир, 1990. 350 с. |
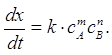 (2)
(2)
|
|
| 17.06.2012 |
| Большое обновление Большой Научной Библиотеки |
| 12.06.2012 |
| Конкурс в самом разгаре не пропустите Новости |
| 08.06.2012 |
| Мы проводим опрос, а также небольшой конкурс |
| 05.06.2012 |
| Сена дизайна и структуры сайта научной библиотеки |
| 04.06.2012 |
| Переезд на новый хостинг |
| 30.05.2012 |
| Работа над улучшением структуры сайта научной библиотеки |
| 27.05.2012 |
| Работа над новым дизайном сайта библиотеки |