|
Курсовая работа: Методы определения хлорид-ионовКурсовая работа: Методы определения хлорид-ионовВведение Защите окружающей среды от возрастающей антропогенной нагрузки в настоящее время уделяется все большее внимание во всем мире. Развитие промышленности, в том числе и химической, увеличение добычи ископаемого сырья, расширение использования транспорта сопровождается поступлением в окружающую среду больших количеств различных загрязняющих веществ. Сильное загрязнение окружающей среды (воды, воздуха, почвы) приводят к возникновению неблагоприятных последствий: нарушению нормальной жизнедеятельности биосферы, изменению климата, исчезновению многих видов растений и животных, ухудшению здоровья населения. Для предотвращения или снижения загрязнения издаются природоохранительные законы и проводятся различные мероприятия – технологические, санитарно-технические, технические, правовые, медицинские и т.п. В основе всех мероприятий лежит контроль за содержанием вредных веществ, который регламентируется санитарно-гигиеническими нормативами – ПДК. Контроль необходим для получения информации об уровне загрязнения, а также об источниках выбросов, причинах и факторах, определяющих загрязнение. Полученные данные позволяют выбирать или проводить защитные, оздоровительные мероприятия и следить за их выполнением. К качеству контроля предъявляются требования надежности и точности, применяемые методы должны быть достаточно чувствительны и избирательны. Независимо от техники выполнения надежность результатов анализа зависит от учета возможных химических, фотохимических и биохимических превращений загрязняющих веществ в разных средах, а также возможности миграции их из одной среды в другую. Данные о загрязнении одной среды должны увязываться с данными о загрязнении другой среды. 1. Распространение хлорид-иона Хлорид-ион образуется в результате растворения и сольватации ионных солей, содержащих анион хлора (хлориды). Следовательно, существование хлорид-иона возможно только в водных растворах. В почвах хлорид ион может также содержатся в составе кристаллических солей. В природе хлор представленный хлорид ионом имеет значительное распространение: 0,02% от массы земной коры. Для сравнения это столько же, сколько и углерода или в 10 раз больше чем свинца. Самые распространенные минералы, содержащие хлорид ион: галит NaCl, сильвинит NaCl*KCl, карналлит KCl*MgCl2. Хлориды тяжелых металлов нерастворимы, хлориды щелочных и щёлочноземельных металлов растворимы все. Значительная растворимость хлоридов обусловила их распространение на планете. Основным местонахождением хлоридов является Мировой океан. По содержанию солей воды мирового океана являются хлоридно-натриевыми. Средняя концентрация хлорид-иона составляет 546 ммоль/л (19 г./л). Значительное содержание хлоридов во внутренних водоёмах явление редкое. Оно колеблется в пределах 5–80 мг/л. Повышенное содержание хлоридов объясняется загрязнением водоема сточными водами некоторых производств. Однако тому причиной может быть и выщелачивание материнской породы содержащей хлоридные соли. Содержание хлорид иона в поверхностных слоях почвах, также не может достигать значительных величин вследствие интенсивного вымывания хлоридов под воздействием атмосферных осадков. Однако возможно присутствие значительных концентраций хлоридов в следующих случаях: – вследствие засоления почв в результате подъёма высокоминерализованных подземных вод; – в результате постоянного притока вод с последующим испарением жидкости. Отсюда два различных местанахождения хлоридов. В первом случае это жидкость влажной почвы, а во втором растворённые хлориды образуют включения кристаллических солей в грунте. 2. Методы определения хлорид-иона 2.1 Общие положения Необходимость определения хлорид-ионов возникает при анализе различных веществ, природных, питьевых и сточных вод. Контроль содержания хлорид-ионов требует различных уровней – от макроконцентраций до 10-7% в особо чистой воде. Существующие государственные стандарты (ГОСТы), регламентируют, какое веществом каким методом и с помощью какого оборудования нужно определять. Современные нормативные документы, регламентирующие процедуру контроля содержания загрязнителей в водах различного происхождения, разрешают использование химических, физико-химических и физических методов анализа. Основная масса лабораторий, проводящих мониторинг вод, не всегда располагает современным оборудованием для реализации физических методов анализа, позволяющих быстро, правильно и точно определять концентрацию загрязнителей. Наиболее массово по-прежнему представлены химические методы. Возможности разработанных на основе этих методов методик определения содержания в воде неорганических загрязнителей не всегда удовлетворяют требованиям ГОСТ, особенно при анализе вод природного происхождения. Многие загрязнители в воде можно обнаруживать разными способами, на разном оборудовании, но разные методы анализа дают различную погрешность, некоторые могут не учитывать какие-либо мешающие факторы. Определение хлоридов в этом отношении имеет ряд преимуществ. Их содержание редко пускается до микроконцентраций, и поэтому основные методики определения хлоридов всё ещ остаются методами «мокрой» химии. Однако в последнее время инструментальные методы применяются все чаще. Инструментальные методы позволяют автоматизировать анализ, сделать его экспрессным, значительно уменьшают расход вспомогательных реактивов. Определение хлоридов можно проводить такими методами: – титриметрия; – потенциометрия; – нефелометрия; – кондуктометрия; Титриметрическое определение хлоридов может выполняться как химическими так и инструментальными методами анализа. 2.2 Химические методы определения хлорид иона 2.2.1 Требования к титриметрическим методам определения Титриметрические определения хлоридов, основаны на реакциях образования осадков малорастворимых соединений. Не все реакции сопровождающиеся выпадением осадков применимы в объемном анализе. В этих реакциях пригодны только некоторые реакции, удовлетворяющие определенным условиям. Реакция должна протекать строго по уравнению и без побочных процессов. Образующийся осадок должен быть практически нерастворимым и выпадать достаточно быстро, без образования пересыщенных растворов. К тому же необходимо иметь возможность определять конечную точку титрования с помощью индикатора. Наконец, явления адсорбции (соосаждения) должны быть выражены при титровании настолько слабо, чтобы результат определения не искажался. Наименования отдельных методов осаждения происходят от названий применяемых растворов. Метод, использующий раствор нитрата серебра, называют аргентометрией. Тиоцианатометрия основана на применении раствора тиоцианата аммония NH4SCN (или калия KSCN) и служит для определения следов С1- в сильнощелочных и кислых растворах. Дорогостоящий аргентометрический метод определения галогенидов по возможности стараются заменять меркурометрическим. В последнем используют раствор нитрата ртути (I) Hg2(NO3)2. 2.2.2 Аргентометрия Объемный аналитический метод, основанный на реакциях осаждения ионов галогенов катионами серебра с образованием малорастворимых галогенидов:
Cl-+Ag+= AgCl↓
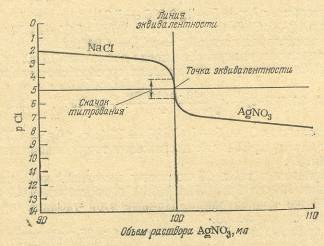
При этом используют раствор нитрата серебра. Если же анализируют вещество на содержание серебра, то пользуются раствором хлорида натрия (или калия). Для понимания метода аргентометрии большое значение имеют кривые титрования. В качестве примера рассмотрим случай титрования 10,00 мл 0,1 н. раствора хлорида натрия 0,1 н. раствором нитрита серебра (без учета изменения объема раствора).
Рисунок 2.1 – Кривая титрования раствора NaCl раствором AgNO3
До начала титрования концентрации хлорид-ионов в растворе равна общей концентрации хлорида натрия, т.е. 0,1 моль/л или [Cl-]=l0-1. Обозначив отрицательный логарифм концентрации (или активности) определяемых хлорид-ионов через рСl, можно написать:
рCl = – lg [Cl-] = – lg l0-1= 1.
Когда к титруемому раствору хлорида натрия будет прибавлено 9,00 мл раствора нитрата серебра и 90% хлорид-ионов будут осаждены, концентрация их в растворе уменьшится в 10 раз и станет равна 10-2 моль/л, а рС1 будет равен 2. Поскольку же величина ПPAgCl=10-10, концентрация ионов серебра при этом составит:
[Ag+] = 10-10/[С1-] = 10-10/10-2 = 10-8 моль/л, или pAg= – lg[Ag+] = – lgl0-8 = 8.
Аналогичным образом вычисляют все остальные точки для построения кривой титрования. В точке эквивалентности pCl=pAg= 5 (см. табл. 2.1).
Таблица 2.1 – Изменение рСl и pAg при титровании 10,00 мл 0,1 н. раствора хлорида натрия 0,1 н. раствором нитрата серебра
Интервал скачка при аргентометрическом титровании зависит от концентрации растворов и от значения произведения растворимости осадка. Чем меньше величина ПР получающегося в результате титрования соединения, тем шире интервал скачка на кривой титрования и тем легче фиксировать конечную точку титрования с помощью индикатора. Наиболее распространено аргентометрическое определение хлора по методу Мора. Сущность его состоит в прямом титровании жидкости раствором нитрата серебра с индикатором хроматом калия до побурения белого осадка. Индикатор метода Мора – раствор К2СгО4 дает с нитратом серебра красный осадок хромата серебра Ag2CrO4, но растворимость осадка (0,65*10-4Э/л) гораздо больше растворимости хлорида серебра (1.25*10-5 Э/л). Поэтому при титровании раствором нитрата серебра в присутствии хромата калия красный осадок хромата серебра появляется лишь после добавления избытка ионов Ag+, когда все хлорид-ионы уже осаждены. При этом всегда к анализируемой жидкости приливают раствор нитрата серебра, а не наоборот. Возможности применения аргентометрии довольно ограничены. Её используют только при титровании нейтральных или слабощелочных растворов (рН от 7 до 10). В кислой среде осадок хромата серебра растворяется. В сильнощелочных растворах нитрат серебра разлагается с выделением нерастворимого оксида Ag2O. Метод непригоден и для анализа растворов, содержащих ион NH4+, так как при этом образуется с катионом Ag+ аммиачный комплекс [Ag(NH3)]+. Анализируемый раствор не должен содержать Ва2+, Sr2+, Pb2+, Bi2+ и других ионов, дающих осадки с хроматом калия. Тем не менее аргентометрия удобна при анализе бесцветных растворов, содержащих С1 – – ионы. Растворы, содержащие Ag+, не выливают в раковину, а собирают в специальную склянку, так как серебро из них регенерируют. Кроме метода Мора при определении хлоридов аргентометрическим титрованием применяется также метод Фаянса. Он основан на прямом титровании растворов содержащих галогенид ионы стандартным раствором AgNO3 в присутствии адсорбционных индикаторов: Ag+ + Cl-→ AgCl mAgCl + n Ag+(изб)→ (AgCl)mAg+n (AgCl)mAgn+ + Ind- → [(AgCl)mAg+n] Ind-
Титрование хлоридов в присутствии флуоресцеина проводят в нейтральной среде. При повышенной концентрации ионов водорода флуоресцеин, являющийся кислотой (HInd), диссоциирует слабо вследствие подавления его диссоциации кислотой. Поэтому концентрация Ind-ионов становится очень малой. В щелочном растворе осаждается Ag2O. В умеренно кислой среде обычно титруют в присутствии других индикаторов: дихлорфлуоресцеина и эозина. Титрование в кислых средах выгодно отличается от титрования в нейтральных растворах, так как дает возможность вести определение в присутствии гидролизующихся солей, разлагающихся водой с образованием осадков гидроокисей и оксихлоридов (Al3+, Fe3+ и др.). В мерную колбу и вносят аликвоту пробы. После чего доводят объем раствора до метки и тщательно перемешивают. Для определения хлорида отбирают аликвотные части исследуемого раствора (по 25 мл), переносят их в конические колбы, прибавляют по 5 капель раствора флуоресцеина и титруют стандартным раствором AgNO3, непрерывно перемешивая. По мере прибавления по каплям раствора AgNO3 титруемая смесь мутнеет. Вблизи точки эквивалентности наблюдается частичная коагуляция коллоидного осадка AgCl. В этот момент титруют еще более внимательно и осторожно, сильно перемешивая содержимое колбы, титрование заканчивают, когда белый осадок AgCl окрашивается в красный цвет. Титрование выполняют 3 – 4 раза и, получив три сходящихся результата, вычисляют результаты анализа. 2.2.3 Роданометрия Роданометрический метод (метод Фольгарда) титриметрического анализа основан на применении в качестве осадителя титрованного раствора, содержащего SCN--ионы:
Ag+ + SCN- → AgSCN
В качестве стандартных растворов используют: для определения ионов роданид аммония; для определения галогенидов и других ионов – нитрат серебра и роданид аммония. Роданометрическим методом пользуются для определения галоген-ионов и серебра в серебряных сплавах. В роданометрии в качестве индикатора для определения точки эквивалентности. применяют насыщенный раствор железо-аммонийных квасцов. Метод Фольгарда обладает рядом достоинств по сравнению с методом Мора. Роданометрический метод применим для определения хлоридов, бромидов, иодидов, роданидов и ионов серебра. Метод применим для титрования кислых растворов так как осадок AgSCN нерастворим в кислотах. Эта особенность метода делает очень удобным при анализе серебряных сплавов, которые растворяют в кислотах, и количественном определении галогенидов в сильнокислых средах, так как галогениды в указанных средах нельзя титровать по методу Мора или в присутствии адсорбционных индикаторов. Другие ионы (Ва2+, РЬ2+ и др.), мешающие определению по методу Мора, в большинстве случаев не мешают определению по методу Фольгарда. Определение С1--ионов по методу Фольгарда основано на применении метода обратного титрования. Хлорид-ионы сначала осаждают определенным объемом стандартного раствора AgNO3, взятого с избытком. Затем оттитровывают не вступивший в реакцию с хлоридом избыток AgNO3 стандартным раствором NH4SCN в присутствии железо-аммонийных квасцов в качестве индикатора. По разности результатов двух титрований определяют объем раствора AgNO3, израсходованного на осаждение С1- Таким образом, последовательно протекают три реакции:
Ag+ + Cl- → AgCl Ag+ + SCN- → AgSCN 3 SCN- + Fe3+ → Fe(SCN)3
Однако в тот момент, когда избыток Ag+ будет оттитрован роданидом, избыток SCN – вступает, кроме того, в реакцию с AgCl:
AgCI+ SCN- ↔ AgSCN + Сl-
Так как роданид серебра (ПPAgSCN = 10-12) менее растворим, чем хлорид серебра (ПPAgCl=. 1,7*10-10), то указанное равновесие сдвигается слева направо. В момент равновесия отношение [C1-]/[SCN-] равно отношению ПPAgCl/ПPAgSCN. Следовательно:
[Cl-]/[SCN-] = ПPAgC/ ПPAgSCN =
т.е. равновесие устанавливается тогда, когда [SCN»] станет в 170 раз меньше [С1-]. В момент равновесия:
[Сl-] =
Следовательно, в точке эквивалентности при избытке SCN- и установившемся равновесии [SCN-] = 1,3*10-5: 170 = 8*10-8 моль/л. Таким образом, равновесие установится тогда, когда практически весь избыток SCN – вступит в реакцию двойного обмена с AgCl. Поэтому конечную точку титрования трудно заметить, так как появившееся розово-красное окрашивание, вызываемое образованием Fe(SCN)3, быстро исчезает вследствие обменной реакции:
Fe(SCN)3 + 3AgCl → Fe3+ + ЗСl- + 3AgSCN
Для предупреждения этой реакции применяют различные способы. Наиболее эффективно отделение осадка AgCl фильтрованием. При этом С1- ионы осаждают избытком раствора AgNO3 в мерной колбе, доводят объем раствора до метки, смесь взбалтывают 5–10 мин и отфильтровывают по частям через сухой фильтр. Первые порции фильтрата отбрасывают. Аликвотные части фильтрата (25 мл из 250 мл) титруют роданидом. Для проведения анализа по методу Фольгарда, берут 100 см3 раствора. Титрованные растворы нитрата серебра и роданида аммония помещают в две различные бюретки. Отбирают из мерной колбы в коническую колбу аликвотное количество раствора, добавляют 2 капли азотной кислоты, 1 мл раствора индикатора и прибавляют отмеренный избыток раствора нитрата серебра (50 мл). Затем приступают к титрованию полученной смеси титрованным раствором роданида аммония до появления красного окрашивания раствора. Определение повторяют до тех пор, пока результаты трех титрований будут расходиться не более чем на 0,05 мл. В случае необходимости выпавший осадок отфильтровывают или добавляют в анализируемый раствор бензол и ведут определение, как указано выше. При известном навыке определение не занимает много времени и приводит к достаточно точным результатам. 2.2.4 Меркуриметрия Метод основан на применении в качестве титранта раствора солей ртути (II). При взаимодействии Hg2+ с хлорид ионами образуется слабо диссоциированное соединение:
Hg2+ + Cl- → [HgCl]+
После достижения точки эквивалентности, в титруемом растворе появляются избыточные Нg2+-ионы, которые обнаруживают при помощи соответствующего индикатора, образующего с Hg2+ характерные соединения. В качестве стандартных растворов для определения галогенидов, цианидов и роданидов применяют нитрат или перхлорат ртути(II), а для определения ионов хорошо диссоциирующих солей ртути – роданид аммония. В меркуриметрии в качестве индикаторов применяют нитропруссид натрия, дающий бесцветный осадок с Hg2+, дифенилкарбазон, образующий синий осадок, р-нитрозо-нафтол, внутрикомплексное соединение которого с Hg2+ красного цвета. И.С. Мустафин и О.В. Сиванова в 1964 г. предложили для этой же цели применять нитрозооксин в смеси с красителем кислотным синим антрахиноновым; последний прибавляется в качестве светофильтра. Такой индикаторный раствор, названный авторами гидрон III, при избытке галогенидов окрашен в зеленый цвет, переходящий в красный при избытке Hg2+. Индикатор позволяет работать с 2,5*10-3 н. раствором Hg2+ и определять, например, 0,03 мг хлоридов в 10 мл титруемого раствора. Меркуриметрический метод широко применяется благодаря многим преимуществам по сравнению с аргентометрическими методами. 1. Меркуриметрический метод позволяет вести прямое определение анионов в кислой среде. 2. Этот метод применяется не только для определения галогенидов, цианидов и роданидов, но и для определения ионов окисной ртути. 3. Многие ионы, мешающие определению по методу Мора и Фольгарда, не оказывают влияния на точность определений с помощью нитрата или перхлората окисной ртути. 4. Соединения ртути являются менее дефицитными, чем соли серебра, и легко регенерируются. Меркуриметрическое определение хлоридов выполняется методом прямого титрования анализируемого раствора раствором нитрата ртути (ІІ) в присутствии индикатора нитропруссида натрия или дифенилкарбазона. Титрование ведётся до появления сине-фиолетового окрашивания. Меркуриметрический метод, равно как и другие методы, основанные на применении солей ртути, имеет весьма существенный недостаток: соли ртути ядовиты, работа с ними требует большой аккуратности и применения необходимых мер предосторожности. 2.2.5 Меркурометрия Меркурометрический метод титриметрического анализа основан на применении титрованных растворов солей ртути(I) (меркуро-ионов). При взаимодействии [Hg2]2+-ионов с хлоридами, бромидами, иодидами и т.д. образуются осадки малорастворимых галогенидов Hg2Cl2, Hg2Br2, Hg2I2, например: [Hg2]2+ + 2Сl- → Hg2Cl2 Меркурометрический метод по сравнению с аргентометрическим дает некоторые преимущества. 1. При меркурометрическом методе не требуется ценных препаратов серебра. 2. Соли ртути (I) менее'растворимы, чем соответствующие соли серебра, и поэтому при титровании хлоридов нитратом ртути(I) наблюдается резкий скачок титрования вблизи точки эквивалентности. 3. Определение меркурометрическим методом можно проводить в кислых растворах методом прямого титрования. Недостатком меркурометрического метода является ядовитость солей ртути. Поэтому при работе с этими солями следует соблюдать большую осторожность. Применение меркурометрического метода при количественных определениях растворимых хлоридов и бромидов пока ограничено. В меркурометрическом методе титрования в качестве индикаторов применяют: Дифенилкарбазон, образующий с [Нg2]2+-ионами осадок синего цвета. Роданид железа Fe(SCN)3. При титровании (например, хлоридов) растворами солей ртути(I) в точке эквивалентности раствор обесцвечивается. Избыток [Hg2]2+-ионов реагирует с Fe(SCN)3 по уравнению: 3 [Hg2]2+ + 2 Fe(SCN)3 → 3Hg2(SCN)2 + 2Fe3+ 2.3 Инструментальные методы определения хлорид-ионов 2.3.1 Нефелометрическое определение хлоридов При прохождении пучка света через дисперсные системы наблюдается рассеяние или поглощение света твердыми частицами. Это явление положено в основу нефелометрии и турбидиметрии. Интенсивность светового потока, рассеиваемого небольшими твердыми частицами взвеси, описывается уравнением Рэлея:
где I и I0 – интенсивности рассеянного и падающего света соответственно; F – функция, зависящая от показателя преломления частиц в растворе; N – общее число частиц во взвеси; V – объем частицы; λ – длина волны падающего света; г – расстояние до наблюдателя; β – угол между направлениями падающего и рассеянного света. При нефелометрических определениях все измерения проводят при определенных значениях F, V, г, р. Поэтому, объединяя их в одну константу, можно записать: I = I0KN = I0KC () Отсюда интенсивность рассеянного светового потока прямо пропорциональна числу частиц во взвесях, т.е. концентрации частиц, находящихся в растворе. Из приведенной выше формулы следует, что интенсивности рассеянного света в двух растворах с частицами одинаковой формы и размеров относятся между собой, как концентрации частиц определяемого вещества:
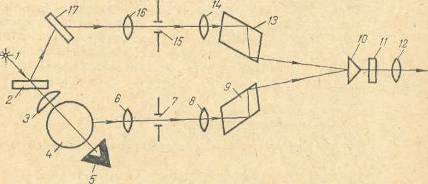
Это уравнение лежит в основе нефелометрических определений. При нефелометрических определениях измеряют интенсивность рассеянного света в направлении, перпендикулярном к направлению первичного пучка света. Турбидиметрические измерения производятся в направлении распространения светового потока. Приведенные уравнения справедливы только для очень разбавленных суспензий (не более 100 мг на 1 л). Турбидиметрические и нефелометрические методы обладают высокой чувствительностью. Однако применяются они не широко, что объясняется трудностью получения взвесей с одинаковыми размерами частиц. Количественные нефелометрические и турбидиметрические определения проводят, пользуясь калибровочной кривой. Для проведения измерений используют прибор – нефелометр. Оптическая схема нефелометра НФМ изображена на рис. 2.2.
Рисунок 2.2 Оптическая схема нефелометра НФМ. Свет от лампы накаливания 1 проходит через стеклянную пластинку 2, конденсор 3 и попадает в кювету 4, помещенную в камеру с дистиллированной водой. Камеру с водой применяют для того, чтобы уменьшить рассеивание света стенками кюветы. Световой поток, прошедший через кювету, гасится в светоловушке 5, а части светового потока, рассеянного частицами взвеси в кювете 4 и стеклянным рассеивателем 17, собираются насадочными линзами 6 и 16. Образовавшиеся два пучка проходят через диафрагмы 7 и 15, связанные с отсчетными барабанами и объективами 8 и 14, направляются в ромбические призмы 9 и 13. Бипризма 10 лает возможность наблюдать в поле зрения окуляра 12 интенсивность двух пучков света. При нефелометрических определениях на пути пучков света вводят светофильтры 11, применение которых нивелирует разницу в оттенках двух световых потоков. Нефелометрическое определение хлорид-ионов основано на реакции осаждения хлоридов нитратом серебра: Ag+ + Cl- → AgCl При малых концентрациях хлорид-ионов выпадение осадка не происходит, а возникает помутнение раствора. Степень помутнения зависит от концентрации хлоридов в растворе. Для стабилизации растворов вводят стабилизирующие компоненты. Анализ проводится следующим образом. Из анализируемого раствора отбирают микропипеткой 5 мл раствора и помещают в мерную колбу емкостью 50 мл. В нее же прибавляют 10 мл 0,1 н. раствора азотной кислоты, 2 мл 0,5%-ного раствора желатины, дистиллированной воды до общего объема приблизительно 30 мл, 10 мл 0,005 М раствора AgCl и доливают водой до метки. Содержимое колбы тщательно перемешивают. Через 5 мин раствор переносят в кювету нефелометра и измеряют рассеивание света не менее 3 раз. Из полученных отсчетов вычисляют среднее значение и по калибровочной кривой определяют содержание хлорид-ионов. Калибровочный график строят следующим образом. Из эталонного раствора КС1 (вводят 0,1 г КС1 в мерную колбу емкостью 500 мл и доводят водой до метки) отбирают в четыре мерные колбы емкостью по 50 мл микропипеткой соответственно 2,0; 4,0; 6,0; 8,0 мл и приготавливают стандартные растворы, добавляя в них все реактивы, указанные выше. Начинают измерения с пробы, имеющей наибольшую концентрацию. Раствор помещают в кювету. Устанавливают светофильтр, цвет которого близок к окраске исследуемого раствора в рассеянном свете. Если жидкость бесцветна, устанавливают зеленый светофильтр. Оба отсчетные барабаны ставят на «0» и подбирают такой рассеиватель, при котором в окуляре левое фотометрическое поле будет несколько светлее правого. Вращением правого барабана уравнивают фотометрические поля по яркости и отсчитывают «кажущуюся» оптическую плотность. 2.3.2 Потенциометрическое определение хлорид ионов Потенциометрические методы анализа делятся на потенциометрическое титрование и прямую потенциометрию. Потенциометрическое титрование преследует чисто прикладную цель количественного определения данного вещества в растворе путем его титрования стандартным раствором соответствующего реагента. При титровании в исследуемый раствор опускают индикаторный электрод, возникновение потенциала на котором обусловливается определяемым веществом непосредственно (если оно электроактивно) или косвенно (если оно неэлектроактивно) в результате химического взаимодействия его с каким-либо другим потенциалопределяющим компонентом. В процессе химической реакции (например, титрования) за изменением концентрации определяемого вещества следят по изменению потенциала индикаторного электрода. Потенциометрическое титрование при прочих равных условиях имеет ряд преимуществ по сравнению с визуальными титриметрическими методами анализа. К применяемым в потенциометрическом титровании химическим реакциям предъявляются те же требования, что и при обычном титриметрическом анализе. В отличие от обычного титриметрического метода, основанного на применении цветных индикаторов, в потенциометрическом методе титрования индикатором является электрод, на котором протекает индикаторная электрохимическая реакция. Скачок потенциала в точке эквивалентности или вблизи нее дает возможность найти конечную точку титрования по кривым титрования или сам скачок принимается как показатель момента завершения реакции. Для определения хлорид ионов применяется метод осадительного титрования. Определение ведётся титрованием раствором с ионами серебра. Индикаторный электрод – серебро металлическое. Химическая реакция выражается уравнением: Ag+ + Cl- → AgCl↓ Разность равновесных потенциалов между двумя точками титрования, отвечающая скачку потенциала, равна: ΔЕ = Е2 – Е1 = Е20 Е10 + υ lg 10-3 = υlg 10-8 – υlg ПPAgСl
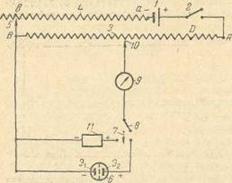
1 – источник постоянного тока с малым выходным напряжением; 2 включатель тока; 3 – делитель напряжения; 4 – переменное сопротивление; 5, 10 – скользящие контакты; 6 – электролитическая ячейка: 7 – переключатель тока; 5 – прерыватель тока; 9 – индикатор тока; 11 – элемент Вестона; Э1 и Э2 – электроды (индикаторный и сравнения). Рисунок 2.3 – Простейшая схема установки для измерения э.д.с. гальванических элементов компенсационным методом при потенциометрическом титровании Измерение потенциала возникающего в цепи измеряется потенциометрами. Почти все приборы для измерения э.д.с. потенциометрической ячейки – потенциометры – имеют следующую схему (рис. 2.3). Один полюс внешнего источника постоянного тока через переключатель неподвижно присоединен к одному из концов (Л) делителя напряжения с равномерным сечением проволоки и с небольшим сопротивлением (10 – 100 ом). Делитель напряжения обычно снабжен шкалой с равномерными 1100 делениями. Другой полюс источника тока присоединен к переменному сопротивлению малой величины, с которым второй конец (В) делителя напряжения соединяется с помощью подвижного контакта. Таким образом, напряжение источника падает на постоянном участке А В и на некотором участке переменного сопротивления ав. Конец В делителя напряжения присоединяют к одному из электродов Э, ячейки, соблюдая при этом полярность соединения, т.е. полюс источника тока и электрод тем же знаком должны быть присоединены к одному и тому же концу делителя. Второй электрод Э2 подключают последовательно через переключатель, прерыватель тока и индикатор тока к подвижному контакту, свободно перемещаемому на делителе напряжения. Дополнительно к концу В делителя напряжения подключают один из полюсов стандартного элемента Вестона (соблюдая тот же порядок полярности соединения, см. выше), другой полюс которого может быть соединен с помощью переключателя с подвижным контактом. Следовательно, при одном положении переключателя замыкается через прерыватель тока цепь, содержащая элемент Вестона, а при другом – цепь, содержащая потенциометрическую ячейку. Элемент Вестона устанавливается для контроля цены одного деления шкалы делителя напряжения (в милливольтах), так как э. д. с. внешних источников тока известна с недостаточной точностью и со временем самопроизвольно падает (источник тока разряжается). Определение С1- в растворе проводят титрованием 0,05 н. стандартным раствором нитрата серебра с серебряным индикаторным электродом и Нас.КЭ сравнения. Э. д. с. потенциометрической ячейки измеряют компенсационным методом. Теоретически скачок наступает несколько раньше точки эквивалентности, но практически точка эквивалентности и конечная точка титрования совпадают. При аргентометрическом титровании недопустимо применение электролитических ключей, заполненных насыщенным раствором хлорида калия; его следует заменить насыщенным раствором KNO3, не реагирующим с Ag+-ионами. Вся предварительная настройка потенциометра, приемы работы и записи результатов аналогичны изложенным выше, но перед началом титрования раствор должен быть приблизительно 10%-ным относительно нитрата бария. Так как система гетерогенна и происходит адсорбция осадком титрующего и титруемого ионов, потенциал устанавливается не быстро, особенно вблизи конечной точки титрования. Поэтому следует ждать достижения более или менее постоянного значения Е. обычно пользуются данными которые за 1 мин изменяются не более чем на 3–5 мВ. Расчет результата определения производится по формуле: mCl- = 2.3.3 Кондуктометрическое определение хлорид ионов Кондуктометрия электрохимический метод анализа, связывающий электропроводность раствора с его составом. Электрическая проводимость растворов обусловлена наличием в них носителей электрического заряда – ионов. Все растворимые соли диссоциируют на ионы, поэтому проводимость ионных раствором значительно выше молекулярных. Для определения хлоридов в объектах окружающей среды прямая кондуктометрия применяться не может. Прямая кондуктометрия заключается в определении электропроводности раствора содержащего определяемый компонент. Линейный характер носит только электропроводность индивидуальных растворов, либо смесей с точно известными концентрациями. Объекты окружающей среды кроме хлорид-ионов, содержат и другие, влияющие на электропроводность раствора. По этой причине для определения хлоридов используется метод кондуктометрического титрования. Кондуктометрическое титрование используется при определении индивидуальных веществ и анализе разнообразных смесей. Точку эквивалентности при кондуктометрическом титровании определяют по изменению электропроводности раствора. Электропроводность измеряют после добавления каждой порции титранта. Зависимость электропроводности раствора от количества добавленного титранта изображают графически. Полученный график называют кривой кондуктометрического титрования. Кондуктометрические кривые имеют излом, соответствующий точке эквивалентности. Кривые подобного типа могут быть использованы для аналитических целей только в том случае, если перед точкой зквивалентности наблюдается линейное изменение проводимости. При титровании следует проводить большое число измерений электропроводности. Для определения точки эквивалентности используют близкие к ней участки кривых. В методе кондуктометрического титрования могут применяться реакции осаждения: Ag+ +NО3- + Na+ + Cl- → ↓AgCI + Na+ + NO3- Изменение состава ионов приводит к изменению электропроводности раствора. Поскольку реакции осаждения часто протекают не мгновенно, измерение сопротивления раствора при титровании следует проводить после достижения постоянной проводимости. При кондуктометрическом титровании необходимо, прежде всего, чтобы излом кондуктометрической кривой позволял устанавливать точку эквивалентности с достаточной точностью. Чем острее угол излома, тем выше точность. Когда угол излома очень тупой, установление точки эквивалентности затруднено. Для кондуктометрического определения хлорид ионов широко применяется титрование нитратом серебра. Однако этот реагент осаждает также Br-, I-, SCN-, СгО4 – С2О42-, тартрат, цитрат и другие анионы. Титрование сопровождается образованием малорастворимых солей серебра. Изменение проводимости растворов при титровании до точки эквивалентности, зависит от сравнительной подвижности осаждаемых анионов и заменяющих их в растворе NO3 ионов. При титровании С1 (λ0= 76,4), Вr – (λ0=78,1), I – (λ0=78,8) и CrO42 – (λ0= 85) проводимость понижается, так как подвижности этих ионов выше подвижности NO3 (λ0=71,5). Однако при титровании SCN – (λ0=57,4), наоборот, происходит небольшое повышение проводимости, так как его подвижность ниже подвижности NO3-. В зависимости от растворимости солей серебра изменяются концентрации титруемых растворов, при которых удается проводить определения с достаточно высокой точностью. Так, титрование хлоридов можно проводить и в очень разбавленных растворах при концентрации С1 – 0,025 мг/мл и меньше. Это титрование используется для определения С1- в питьевой воде. Между тем I – можно титровать, в растворах, концентрация которых больше 0,005 н., а цитраты только при концентрации не ниже 0,1 н.
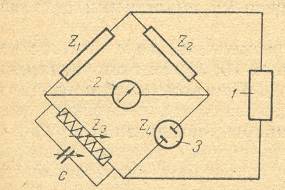
Z1, Z2, Z3, Z4 – плечи мостика, С переменная ёмкость, 1 – генератор звуковой частоты, 2 – гальванометр, 3 электролитическая ячейка. Рисунок 2.4 Мостик Уитстона Электропроводность растворов может быть найдена, если измерить сопротивление электролитической ячейки. Для измерения сопротивления используют переменный ток звуковой частоты, так как постоянный вызывает разложение веществ находящихся в растворе. Сопротивление раствора определяют путём сравнения с эталонным сопротивлением. Для этого служит мостик Уитстона (рисунок 2.4). Сопротивления Z1, Z2, Z3, Z4 можно подобрать так, чтобы ток в диагонали моста отсутствовал, т.е. сопротивления каждой ветви были пропорциональны. Измеряемое сопротивление определяют по формуле: Z4= Где Z – сопротивления соответствующих плечей мостика. 3. Анализы объектов на содержание хлорид-иона 1 Определение массовой доли хлоридов в отложениях парового оборудования электростанций Навеску отложений от 0,5 до 0,8 г, взвешенную на аналитических весах, помещают в химический стакан. Обрабатывают материал 20–30 см3 горячей дистиллированной воды, кислоты и отфильтровывают не растворившийся осадок на плотный беззольный фильтр, собирая фильтрат в мерную колбу вместимостью 100 или 250 см3. Осадок на фильтре промывают горячей дистиллированной водой, собирая промывные воды в ту же колбу. Промывание ведут до исчезновения в фильтрате положительной реакции на присутствие хлорид-иона (проба с AgNO3: несколько капель фильтрата смешивают с раствором, содержащим 1% AgNO3). Из мерной колбы, содержащей фильтрат отбирают пипеткой от 10 до 100 см3 жидкости в зависимости от предполагаемого содержания хлоридов в анализируемом материале. Аликвоту осветлившегося раствора, отобранную пипеткой, помещают в коническую колбу, приливают 2 см3 концентрированной азотной кислоты и дистиллированной воды до общего объема примерно 100 см3. Затем вводят на кончике ножа несколько кристаллов нитропруссида натрия и три капли раствора метилоранжа. Кристаллы нитропруссида натрия растворяют и титруют окрашенную в оранжево-красный цвет жидкость раствором нитрата ртути II такой концентрации, чтобы каждый миллилитр раствора отвечал 1 мг хлор-иона. Титрование ведут до появления мути в жидкости, которая хорошо заметна при прибавлении одной капли ртутного раствора на фоне черной глянцевой бумаги. Массовую долю хлоридов (%) в пересчете на хлористый натрий получают по формуле:
где А – расход титранта, т.е. раствора азотнокислой ртути, см3; Т – содержание хлор-ионов, которому соответствует 1 см3 ртутного раствора, мг/см3; 58,443 и 35,453 – молекулярные веса хлористого натрия и хлор-иона. П – объём аликвотной порции, отобранной для определения, см3; G – навеска, г; В-объем мерной колбы, в которую собран фильтрат после отделения кремнекислоты, см3. 2 Определение ионов хлора в нефтяном буровом растворе Данный тест предназначен для измерения суммарной концентрации растворимых хлоридов в буровом растворе. Источниками ионов хлора в растворе служат хлористый натрий, хлористый кальций и хлористый калий. Для получения правильных результатов титрования фильтрат должен иметь слегка щелочную реакцию рН ≈ 8,3. В процессе титрования одновременно протекают две химические реакции. 1. Аg+ + Сl-→AgCl 2. 2Аg+ + СгО42-→ Аg2СгO4 Результатом первой реакции (образование хлористого серебра) служит появление белых вкраплений или молочное окрашивание раствора. Образование красного хромата серебра начинается лишь после того, как все ионы хлора оказываются связанными в хлористое серебро. После этого нитрат серебра вступает в реакцию с индикаторным раствором хромата калия, в результате чего образуется хромат серебра. Таким образом, для нормального протекания обеих реакций фильтрат должен иметь слабощелочную реакцию, (рН = 8.3). При высоком рН происходит выпадение осадка окиси серебра. Оборудование Шприц со стеклянным наконечником, 5 мл; мерный стакан, 400 мл, стеклянный; магнитная мешалка с «микромешалками» Реактивы: Дистиллированная вода, раствор хромата калия, индикаторный раствор фенолфталеина, раствор серной кислоты 0,1 N, раствор нитрата серебра, растворитель Arcosolv PNP. Методика определения Для начала определите щелочность цельного бурового раствора: 1. Внесите 100 мл растворителя Arcosolv PNP в 400-мл мерный стакан. 2. Наберите в 5-мл шприц не менее 3 мл цельного бурового раствора и введите 2 мл в мерный стакан. 3. Взбалтывайте смесь круговыми движениями до достижения однородности. 4. Добавьте 200 мл дистиллированной воды. Добавьте 15 капель индикаторного раствора фенолфталеина. 5. Перемешивая смесь магнитной мешалкой, медленно титруйте 0,1N серной кислотой до начала исчезновения розовой окраски. Продолжайте перемешивать смесь в течение еще одной минуты и, если розовая окраска не появится вновь, прекратите перемешивание. Иногда бывает необходимо прекратить перемешивание, чтобы обеспечить разделение обеих фаз и возможность более четкого визуального определения цвета водной фазы. 6. Оставьте образец на 5 минут, и если розовая окраска не появится вновь, то это будет свидетельствовать о достижении конечной точки титрования. В случае возобновления розовой окраски проведите повторное титрование серной кислотой. Если розовая окраска окончательно не исчезает, оттитруйте смесь в третий раз, однако в случае возобновления окраски после третьего титрования следует прекратить дальнейшие попытки и принять полученный результат за конечную точку. 7. Закончив определение щелочности, обеспечьте наличие кислой реакции (рН < 7) у смеси, которую предполагается титровать на содержание хлоридов, путем добавления 10–20 капель 0,1 Н серной кислоты. 8. Добавьте 3,0 мл индикаторного раствора хромата калия. 9. Перемешивая смесь с помощью магнитной мешалки, медленно титруйте ее 0.282N раствором нитрата серебра до появления оранжево-розовой окраски, сохраняющейся на протяжении как минимум одной минуты. Иногда бывает необходимо прекратить перемешивание, чтобы обеспечить разделение обеих фаз и возможность более четкого визуального определения цвета водной фазы. 10. Расчет содержания хлоридов в цельном буровом растворе основывается на суммарном количестве миллилитров 0.282N нитрата серебра, потребовавшемся для достижения конечной точки титрования. Расчет содержания хлоридов осуществляется по формуле:
3 Методика количественного химического анализа мясных продуктов на содержание хлоридов методом ионной хроматографии Проба экстрагируется по ГОСТ 9957–73 дистиллированной водой. Отделение анионов хлоридов производится хроматографически на ионообменной колонке с последующим кондуктометрическим детектированием. Относительная погрешность определения хлорид-иона в диапазоне 1–5% составляет 0.1%(2% отн.). Оборудование: хроматограф жидкостный ионный «Цвет-3006М» ТУ 5Е1.550.186 или другой жидкостный ионный хроматограф; весы аналитические ВЛА-200г-М, ГОСТ 24104–89; колбы мерные на 50, 100, 200 и 1000 см3, ГОСТ 1770–74; пипетки мерные вместимостью 1, 2, 5, 10 и 25 см3, ГОСТ 20292–74; колбы конические вместимостью 100 или 200 см3, ГОСТ 10394–72; электромясорубка бытовая по ГОСТ 20469–81; насос вакуумный типаВМ-461; водяная баня; воронки стеклянные, ГОСТ 8613–75; фильтры беззольные бумажные; стакан химический вместимостью 200–250 мл по ГОСТ 10394–72. Реактивы и материалы. 1. Калий хлористый, х.ч., по ГОСТ 4234–77. 2. Сорбент катионообменный КРС-8п ТУ 6–09–10–151–79. 3. Натрий углекислый кислый, хч, по ГОСТ 4201–79. 4. Натрий углекислый, хч, по ГОСТ 83–79. 5. Кислота азотная, хч, по ГОСТ 4461–77. 6. Вода бидистиллированная, ГОСТ 6709072 или деионизованная. 7. Сорбент анионообменный КанК-Аст ВТУ 881202–89. Подготовка проб к анализу Отобранный образец измельчается мясорубкой. 5 г измельченной средней пробы взвешивают в химическом стакане с погрешностью + 0,01 г. и добавляют 200 мл дистиллированной воды. Через 40 мин настаивания (при периодическом помешивании стеклянной палочкой) водную вытяжку фильтруют через бумажный фильтр. Для определения хлоридов берут 1 мл водной вытяжки в мерную колбу на 100 мл и доводят до метки дистиллированной водой. Полученный раствор используют для ввода в хроматограф. Выполнение измерений Готовят градуировочные растворы объемно-весовым методом. Для приготовления исходного раствора хлорид-иона концентрацией 1 г/л необходимо взять навеску 2,1 г хлористого калия и растворить в дистиллированной воде в отдельной мерной колбе вместимостью 1000 мл. Установку и включение хроматографа осуществляют в соответствии с инструкциями по эксплуатации хроматографа Цвет-3006М. На насосе задают расход элюента 2,0 см3/мин, шкалу чувствительности устанавливают «32». Устанавливают режимные параметры и вводят градуировочный раствор в дозатор хроматографа. Снимают хроматограммы стандартных растворов. Градуировочный коэффициент К для хлорид ионов рассчитывается по формуле:
где hгр – высота пика компонента в градуировочном растворе, мм; Сгр – концентрация компонента в градуировочном растворе, мг/л. Проведение анализа Вводят в хроматограф водную вытяжку для определения хлорид-иона. Снимают хроматограмму. Время выхода пика хлорида 10 мин. Концентрацию хлорид-иона в пробе (Спробы) вычисляют по формуле: Спробы(мг/кг)= где hпр – высота пика компонента в пробе, мм; М – навеска пробы, г. 4 Аргентометрическое определение хлоридов в воде Метод основан на титровании хлорид-ионов раствором нитрата серебра в нейтральной или слабощелочной среде (рН=6,5–10) в присутствии индикатора K2CrO4. Сl- + Ag+ → AgCl ↓ Определению мешает присутствие ионов РО43 – (> 25 мг/л), Fe3+ (> 10 мг/л) и катионов, образующих осадки с Сl –. Рабочий раствор нитрата серебра готовят следующим образом: AgNO3 – 2,4 г AgNO3 помещают в мерную колбу на 1 литр и доводят дистиллированной водой до метки. Точную концентрацию устанавливают по раствору NaCl, приготовленному из фиксанала или по точной навеске. Раствор AgNO3 для качественного определения хлорид-ионов -10%-й. Раствор K2CrO4 – 5%-й. Выполнение определения К 5 мл исследуемой воды добавляют 3 капли 10%-го раствора AgNO3. Приближенное содержание Сl – определяют в соответствии с таблицей Таблица 1 Характер осадка хлорида серебра в зависимости от концентрации хлорид-ионов
В зависимости от содержания СГ необходимо брать для анализа различный объем воды. Если содержание хлорид-иона – в пределах 100–250 мг/л, для анализа необходимо брать 100 мл воды; если 250–280 мг/л – 50 мл воды; если > 800 мг/л – объем воды берется < 50 мл.Количественное определение Сl –. К необходимой для анализа аликвоте воды (100 мл или меньше) добавляют 1 мл 5%-го раствора K2CrO4 и титруют раствором AgNO3 до появления оранжево-бурой окраски. Расчет содержания хлоридов ведут по формуле:
где mCl− – масса хлорид-иона, г; CCl − концентрация хлорид-иона, мг/л; ТAgNO3/ Cl- – титр раствора AgNO3 пo хлорид-иону, г/мл; VAgNO3 – объем раствора AgNO3, израсходованный на титрование, мл; VH2O - объем воды, взятой для анализа, л. 5. Меркуриметрическое определение хлоридов Метод основан на титровании хлорид-ионов раствором нитрата ртути (II) Hg(NO3) 2 в кислой среде в присутствии индикатора дифенилкарбазона. 2С1 +Hg2+ →HgCl Метод позволяет определить от 1 до 10 мкг Сl - в пробе. Определению Сl – мешает присутствие ионов S2−, Br−, I-. Рабочий раствор Hg(NO3)2 (0,1 н). Нитрат ртути (II) гигроскопичен, поэтому стандартный раствор его нельзя приготовить растворением точной навески. Обычно готовят вначале раствор Hg(NO3) 2 примерно 0,1 н. Для этого 16,7 г Hg(NO3) 2· 1/2H2О переносят в мерную колбу на 1 литр, добавляют 20 мл 6 н HNO3 (для улучшения растворимости Hg(NO3)2 и предупреждения ее гидролиза) и доводят дистиллированной водой до метки. Затем устанавливают его титр по раствору NaCl. Раствор NaCl – 0,1 н (готовят из фиксанала или по точной навеске). Дифенилкарбазон – 2%-й раствор. Определение титра рабочего раствора Hg(NO3)2. К 10 мл 0,1 н раствора NaСl добавляют 1 мл 2%-го раствора дифенилкарбазона и титруют раствором Hg(NO3) 2 до появления синего окрашивания. Определяют титр раствора Hg(NO3)2
Выполнение определения К 100 мл предназначенной для анализа воды (или к меньшему объему) добавляют 10 мл 2 н раствора HNO3, 1 мл 2%-ного раствора дифенилкарбазона и титруют раствором Hg(NO3)2 до появления сине-фиолетового окрашивания. По результатам титрования рассчитывают содержание хлоридов:
где тСl – масса хлорид-иона, г; СCl – концентрация хлорид-иона, мг/л; ТHg(NO3)2 – титр раствора Hg(NO3)2 по хлорид-иону, г/мл; VHg(NO3)2 – объем раствора Hg(NO3)2, израсходованный на титрование, мл; VH2O – объем воды, взятый для анализа, л. Выводы Определения хлорид-ионов основаны на реакциях осаждения. Изменения концентрации и формы нахождения хлорид ионов вызывает изменение свойств системы, которые регистрируются как аналитический сигнал. |
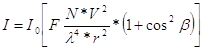 ()
()
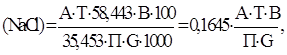 (.1)
(.1)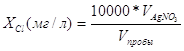 (.2)
(.2)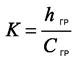 (.4)
(.4)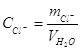 (.7)
(.7)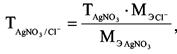 (.8)
(.8)
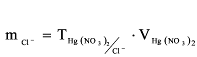
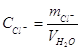 (.)
(.)|
|
| 17.06.2012 |
| Большое обновление Большой Научной Библиотеки |
| 12.06.2012 |
| Конкурс в самом разгаре не пропустите Новости |
| 08.06.2012 |
| Мы проводим опрос, а также небольшой конкурс |
| 05.06.2012 |
| Сена дизайна и структуры сайта научной библиотеки |
| 04.06.2012 |
| Переезд на новый хостинг |
| 30.05.2012 |
| Работа над улучшением структуры сайта научной библиотеки |
| 27.05.2012 |
| Работа над новым дизайном сайта библиотеки |